(UroToday.com) The 2024 Advanced Prostate Cancer Consensus Conference (APCCC) held in Lugano, Switzerland was host to an advanced prostate cancer session. Dr. Himisha Beltran discussed how to identify aggressive variant prostate cancer.
Castrate-resistant prostate cancer is a heterogenous disease with numerous:
- Genomic subtypes
- Examples: BRCA2, microsatellite instability-high (MSI-H), PTEN loss, TP53, and RB1
- Phenotypic variants
- Histologic (e.g., neuroendocrine prostate cancer)
- PSMA-negative/FDG PET avid
- Gene expression
- Resistance mechanisms
- Most are androgen receptor driven (AR mutations/amplifications)
- Non-androgen receptor driven (loss of AR expression/signaling)
This is reflected in the mCRPC clinical phenotype spectrum, whereby some patients have significantly more aggressive disease than others.

This variability spectrum has important therapeutic implications, whereby therapy should be intensified for patients with the most aggressive disease features. We need to develop rational combination strategies that, ideally, should target both AR and non-AR driven disease.
What is aggressive variant prostate cancer (AVPC)? The term AVPC was used for specific clinical criteria in therapy intensification (platinum-taxane) phase 2 clinical trials:
- Exclusive visceral metastases;
- Lytic bone metastases;
- Bulky lymphadenopathy (≥5 cm) or mass (≥5 cm, Gleason ≥8) in prostate/pelvis
- Low PSA (≤10 ng/mL) at initial presentation (before ADT) or at the time of symptomatic CRPC plus high volume (≥20) bone metastases;
- Serum CEA and/or LDH twice the upper limit of normal;
- Short interval (≤6 months) to CRPC;
- Small cell/neuroendocrine carcinoma morphology.
In prostate cancer, therapeutic pressure may lead to small cell carcinoma/neuroendocrine prostate cancer transformation.
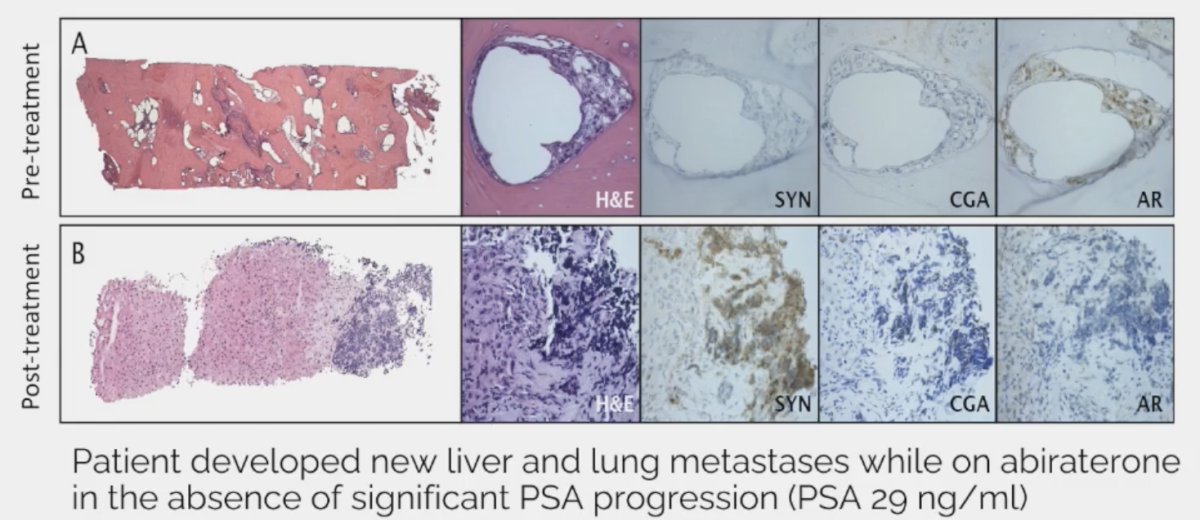
Other notable aggressive features that are not included in the AVPC definition include:
- PSMA negative/FDG avid disease on PET scan
- High ctDNA fraction or circulating tumor cell count - reflective of tumor burden
- Germline and somatic DNA alterations (BRCA2, RB1)
- Liver metastases regardless of other sites
- Parenchymal brain metastases (rare but increasingly reported)
- The list of prognostic biomarkers keeps growing and may inform treatment intensification studies.
Do AVPC clinical features necessarily reflect the underlying biology? In phase 2 platinum trials, AVPC has been defined using clinical parameters. Genomic analysis has demonstrated the following underlying mutations:
- ~50-60% RB1 (by IHC or NGS)
- ~80% TP53
- ~50% PTEN
These mutations appear to be of predictive value, whereby patients with ≥2/3 alterations in TP53, RB1, and PTEN had longer median progression-free (7.5 versus 1.7 months) and overall survival (20.2 versus 8.5 months), compared to those with 0–1 alterations when treated with combination cabazitaxel and carboplatin.1 As such, Dr. Beltran proposed whether loss of 2/3 such tumor suppressors should be used to define AVPC instead of clinical criteria.
Dr. Beltran noted that the most common tumor suppressors lost in CRPC are as follows:
- RB1: 20% versus 3% for primary
- TP53: 45% versus 15 for primary
- PTEN: 40% versus 20% primary
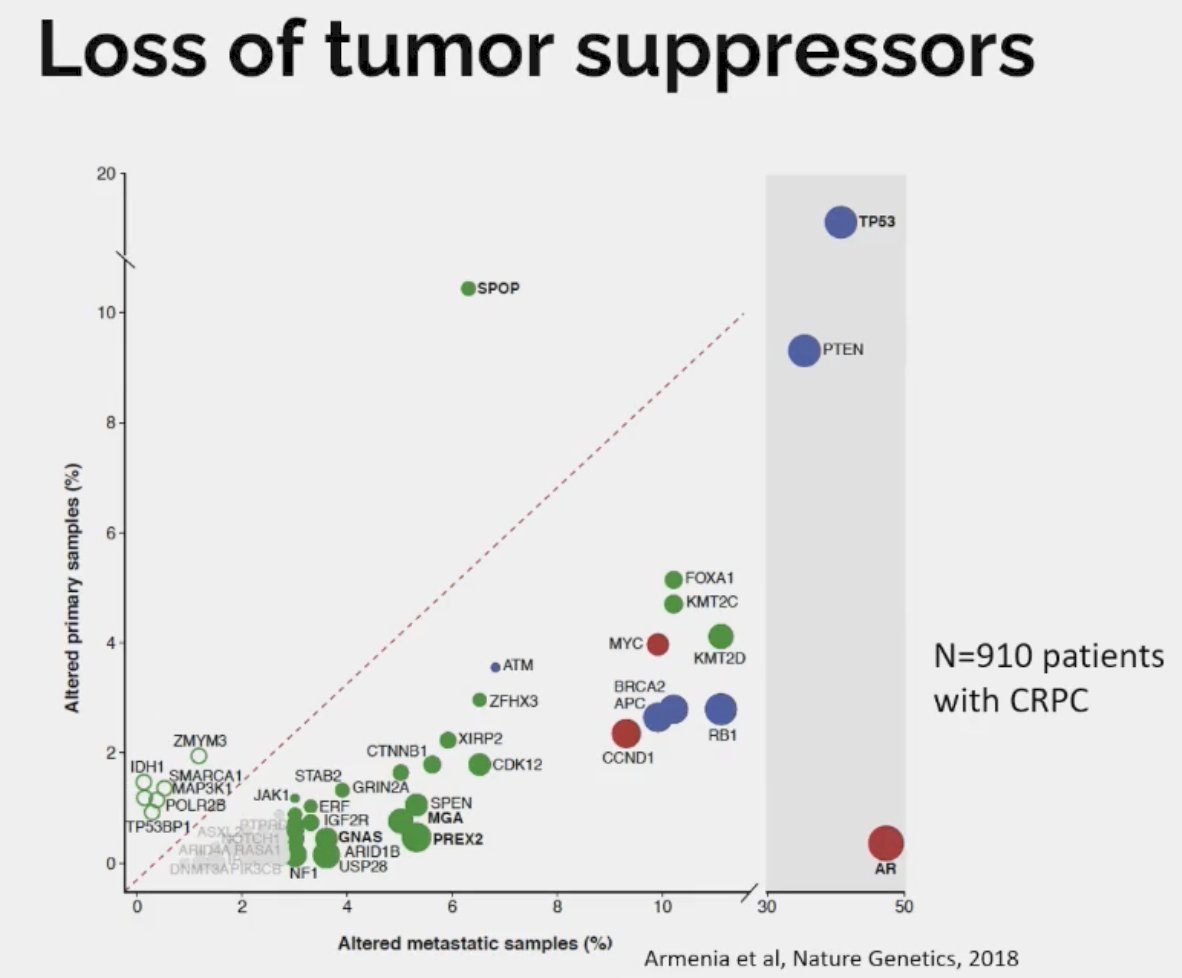
Loss of tumor suppressors is associated with lineage plasticity and AR independence. Drawing parallels to small cell lung cancer (SCLC), it appears that loss of RB1 and TP53 precedes histologic transformation to SCLC. 5–15% of lung adenocarcinomas transform into small cell lung cancer at time of resistance to EGFR inhibitor therapy. Multiple studies have shown that RB1 and TP53 loss detected in lung adenocarcinoma predicts future transformation to small cell lung cancer.
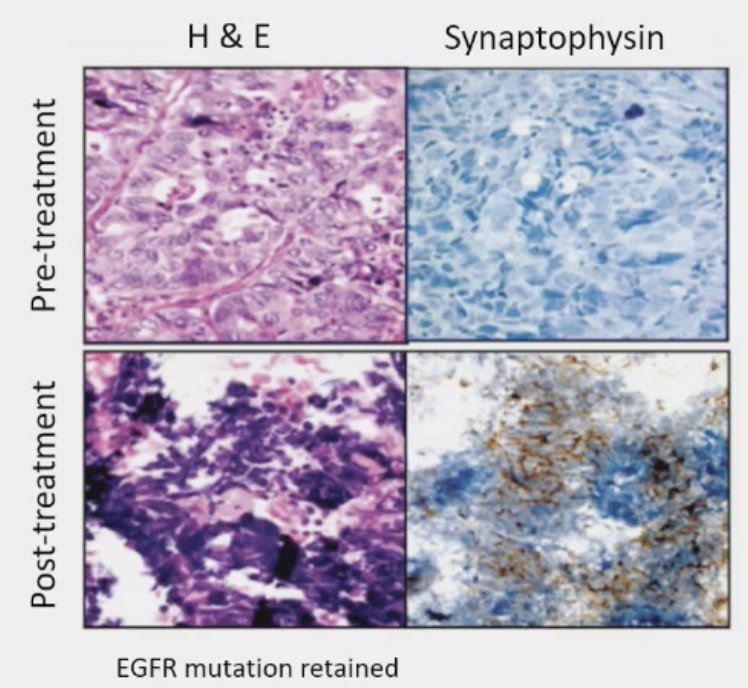
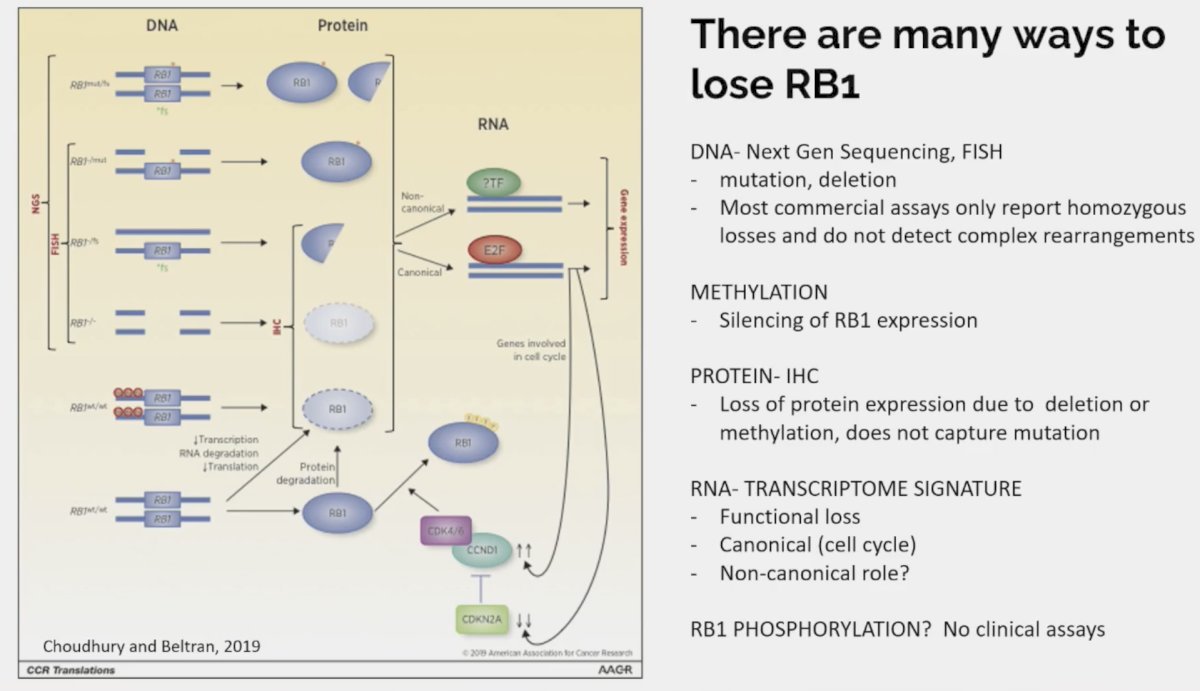
How/what should we measure loss of tumor suppressors in the clinic? Potential candidates, with notable limitations, include:
- Primary tumor: Misses alterations that are acquired at later stages
- Metastatic tumor: At what time point and which lesion/locations should be sampled?
- ctDNA: Has important limitations for detection of numerous deletions
- Immunohistochemistry: What antibodies and cut-offs are most reliable
- Genomic sequencing: Targeted next generation sequencing is the most common but does not detect functional or epigenetic losses.
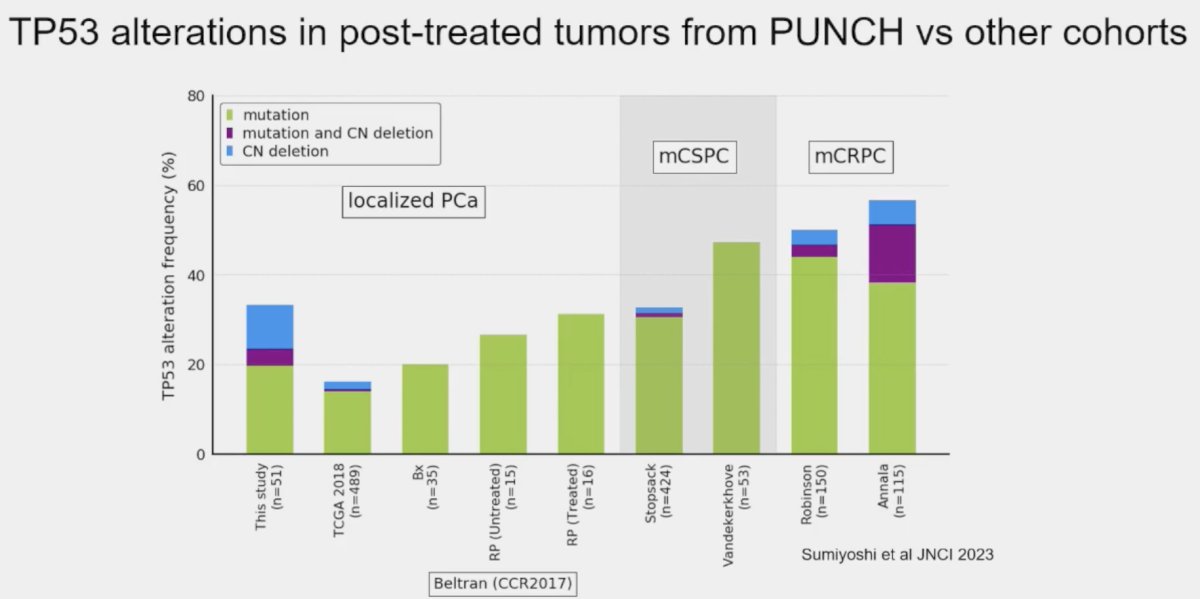
The prevalence of these mutations also appears to be influenced by prior therapy received, even in the neoadjuvant setting. Post hoc analysis of the PUNCH CALGB 90203 (Alliance) phase 3 trial of neoadjuvant chemohormonal therapy prior to radical prostatectomy in high-risk prostate cancer patients demonstrated that TP53 alterations were enriched and associated with shorter overall survival, whereas SPOP alterations were infrequently detected after chemohormonal therapy.2
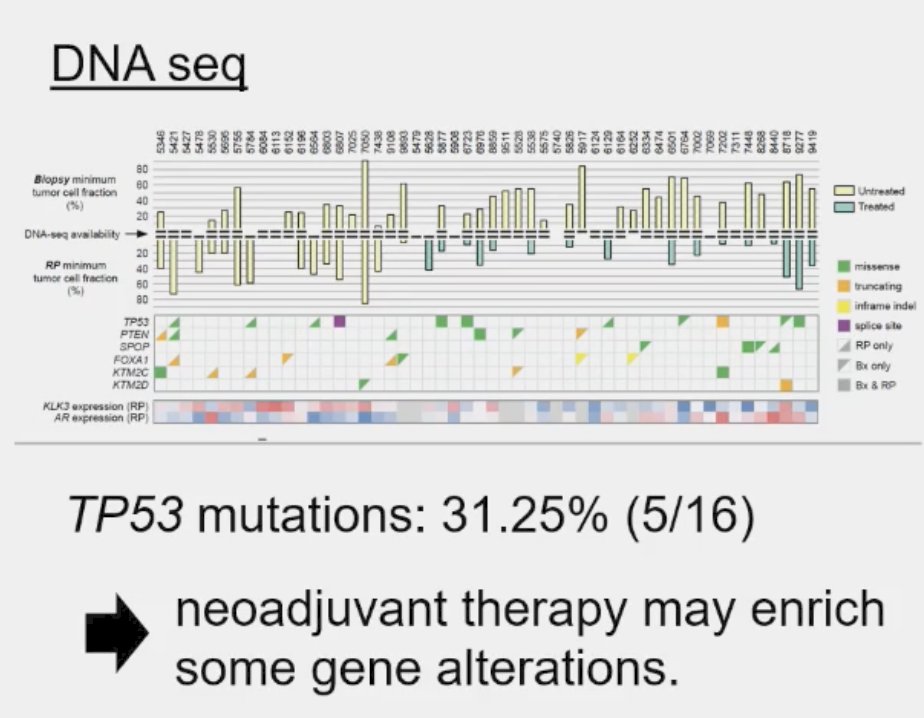
Residual tumors treated with ADT plus docetaxel had mainly TP53 defects and PTEN alterations. Alteration frequency of TP53 tended to be closer to metastatic frequencies.
In contrast to TP53 and PTEN mutations that may occur early and selected for, RB1 alterations are often acquired. In a study published by Rodrigues et al. in 2019, 70 samples from 41 patients (including 20 matched, same patient HSPC-CRPC pairs) underwent fluorescence in situ hybridization (FISH) assessment, and demonstrated the following:
- 20 HSPC samples: RB1 deletions present in 35%
- 20 CRPC samples: RB1 deletions present in 65%
- Of the 13 HSPC samples without deletion, 7 acquired deletions in the CRPC sample
- This suggests heterozygous to monozygous loss with disease progression (note: only homozygous losses are reported on most assays)
Dr. Beltran highlighted the numerous ways in which patients may lose RB1 expression:
What is the ideal method to evaluate for tumor suppressor loss? In 2023, Viscuse et al. evaluated 49 men with mCRPC progression who were undergoing biopsies for standard molecular profiling. TP53, RB1, and PTEN expression were assessed by immunohistochemistry, and patients concurrently underwent next generation sequencing and ctDNA testing. Immunohistochemistry outperformed the other two modalities for detection of tumor suppressor alterations (27%). Notably, immunohistochemistry offers the additional advantages of being cost-effective with specific/validated antibodies and easier to implement across centers.

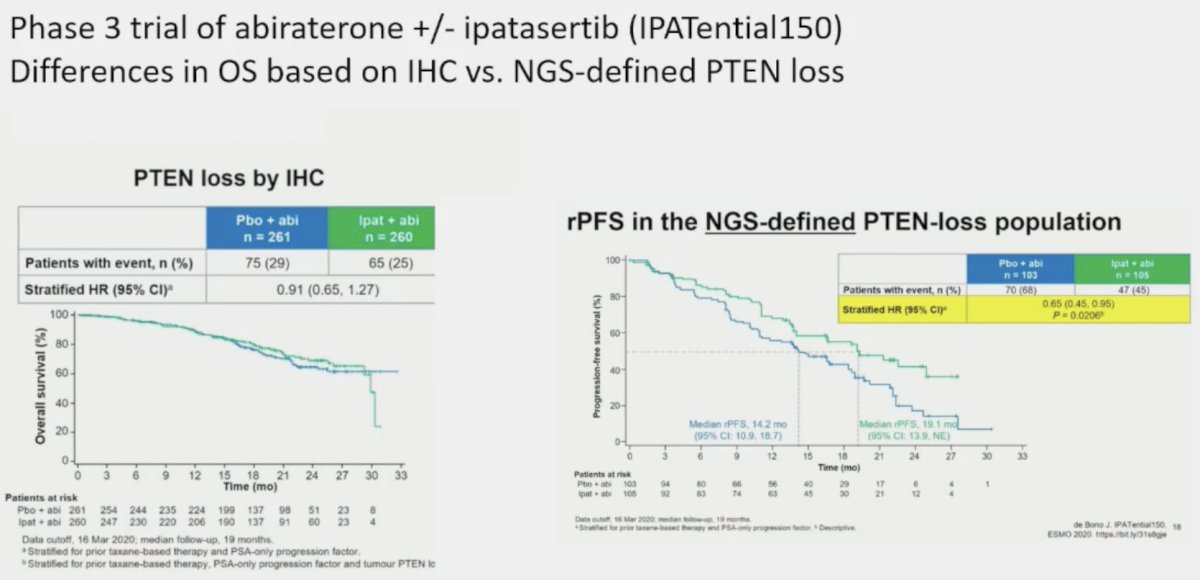
However, p53 immunohistochemistry can be high with mutations but low with deletions. Additionally, issues with regards to quantifying heterogeneity of expression and standardization of testing/reporting remain. This is well-exemplified by an analysis of IPATential 150, a phase 3 trial of abiraterone +/- ipatasertib in patients with PTEN loss. In patients with PTEN loss by immunohistochemistry, there was no radiographic progression-free survival (rPFS) benefit to adding ipatasertib. Conversely, among patients with NGS-defined PTEN loss, adding ipatasertib significantly improved rPFS. Dr. Beltran noted that the lesson here is that multiple assays may be required to provide a full picture of tumor suppressor gene alterations.
Dr. Beltran noted that there is currently no indication to test for tumor suppressor loss in the clinic. However, such tumor mutations are often reported in NGS studies and sometimes clinical immunohistochemistry, indicating likely tumor aggressiveness. This may have potential future implications for:
- Therapy intensification, potentially with platinum agent
- Prompt imaging, potentially with FDG PET
- Biopsy: to evaluate for neuroendocrine prostate cancer
- Correlate with targets (epigenetic, cell surface)
Next, Dr. Beltran addressed neuroendocrine prostate cancer noting that it is an aggressive histologic variant that can arise de novo or after therapy for castrate-resistant prostate cancer. These tumors clinically, pathologically, and molecularly overlap with small cell lung cancer. There is some controversy with regards to its prevalence with some reporting having never or rarely seen this variant, whereas others report proportions as high as 15-20% of CRPC tumors. Possible explanations for this discrepancy include:
- Repeat biopsies are not standardly/routinely done in mCRPC
- NCCN currently recommends considering a biopsy to evaluate for neuroendocrine differentiation
- Variability among pathologists – evaluation and nomenclature is not standardized
- Intra-patient heterogeneity

There is evidence that neuroendocrine prostate cancer arises as a result of lineage plasticity, as illustrated below. While these prostate cancer cells are initially highly PSMA expressing in the adenocarcinoma state, they become low/variable PSMA expressing in the neuroendocrine phase. However, expression of other cell surface molecules such as DLL3 increases, facilitating development of novel imaging and treatment targets.
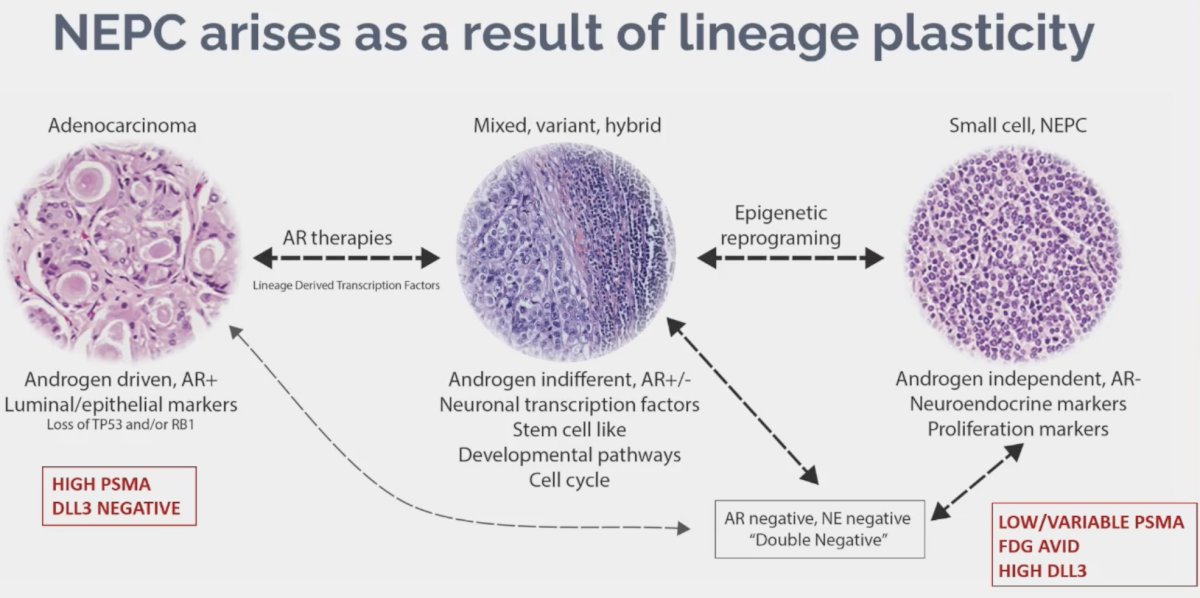
What are the PET features of neuroendocrine prostate cancer? These tumors are often PSMA negative/low, reflecting the lack of androgen receptor expression. This is not universal among all such lesions, as up to 20% of neuroendocrine prostate cancers can express PSMA. Notably, not all PSMA negative lesions are neuroendocrine prostate cancers. These lesions are FDG avid, reflecting metabolic activity; however, this is not specific to neuroendocrine prostate cancer. PET DOTATE has been evaluated in these patients, which performs well for well differentiated neuroendocrine tumors, but poorly for high grade neuroendocrine carcinomas. There are ongoing studies evaluating DLL3 PET in this disease space.
Emerging neuroendocrine prostate cancer biomarkers include:
- Molecular imaging (DLL3 PET, others)
- Liquid biopsies (cDNA methylation, fragment omics, ChiPseq. CTCs)
- Transcriptomic signatures (NEPC score – mRNA based)


Dr. Beltran concluded her presentation as follows:
- Aggressive variants of CRPC exist and may benefit from alternative treatment strategies.
- The current diagnosis of aggressive disease is imperfect and inconsistent and may encompass multiple biologic subtypes.
- Number of emerging diagnostic tools have been developed:
- Clinical criteria (AVPC)
- Tumor suppressor loss (RB1, TP53. PTEN)
- AR gain/mutation/splice variants
- Biopsy (morphology, immunohistochemistry)
- PET (PSMA, FDG)
- Liquid biopsies (cfDNA)
- Standardization of biomarkers in clinical studies will be essential for helping us understand their therapeutic relevance.
Presented by: Himisha Beltran, MD, Associate Professor of Medicine, Department of Medical Oncology in the Lank Division of Genitourinary Oncology and the Division of Molecular and Cellular Oncology at Dana Farber Cancer Institute and Harvard Medical School, Boston, MA
Written by: Rashid Sayyid, MD, MSc – Society of Urologic Oncology (SUO) Clinical Fellow at The University of Toronto, @rksayyid on Twitter during the 2024 Advanced Prostate Cancer Consensus Conference (APCCC) Meeting, Lugano, Switzerland, Thurs, Apr 25 - Sat, Apr 27, 2024.
References:
- Aparicio AM, Shen L, Tapia ELN, et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin Cancer Res. 2016;22(6): 1520-30.
- Sumiyoshi T, Wang X, Warner EW, et al. Molecular features of prostate cancer after neoadjuvant therapy in the phase 3 CALGB 90203 trial. J Natl Cancer Inst. 2024;116(1): 115-26.
- Rodrigues DN, Casiraghi N, Romanel A, et al. RB1 Heterogeneity in Advanced Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2019;25(2): 687-97.
Related Content: