(UroToday.com) The 2024 American Society of Clinical Oncology (ASCO) Annual Meeting held in Chicago, IL was host to a kidney and bladder cancers poster session. Dr. Maria Bourlon presented the safety and patient-reported outcomes of the randomized phase 3 CheckMate 67T trial of subcutaneous versus intravenous nivolumab in patients with previously treated advanced or metastatic clear cell renal cell carcinoma (RCC).
The anti-programmed death 1 (PD-1) receptor antibody nivolumab administered via an intravenous infusion has improved clinical outcomes in multiple tumor types; however, the evolving treatment paradigm has created an unmet need for novel administration options that may improve the patient treatment experience and help reduce inefficiencies in healthcare systems. Subcutaneous administration has several advantages over intravenous infusion, including improved healthcare resource utilization (by decreasing preparation time and length of chair occupancy), and reduced administrative burden, and is preferred by patients. Subcutaneous nivolumab is co-formulated with the enzyme recombinant human hyaluronidase PH20 (rHuPH20), which degrades hyaluronan when injected into the subcutaneous space, enabling the delivery of larger drug volumes, and reduces the number of individual injections required compared with subcutaneous administration without rHuPH20.
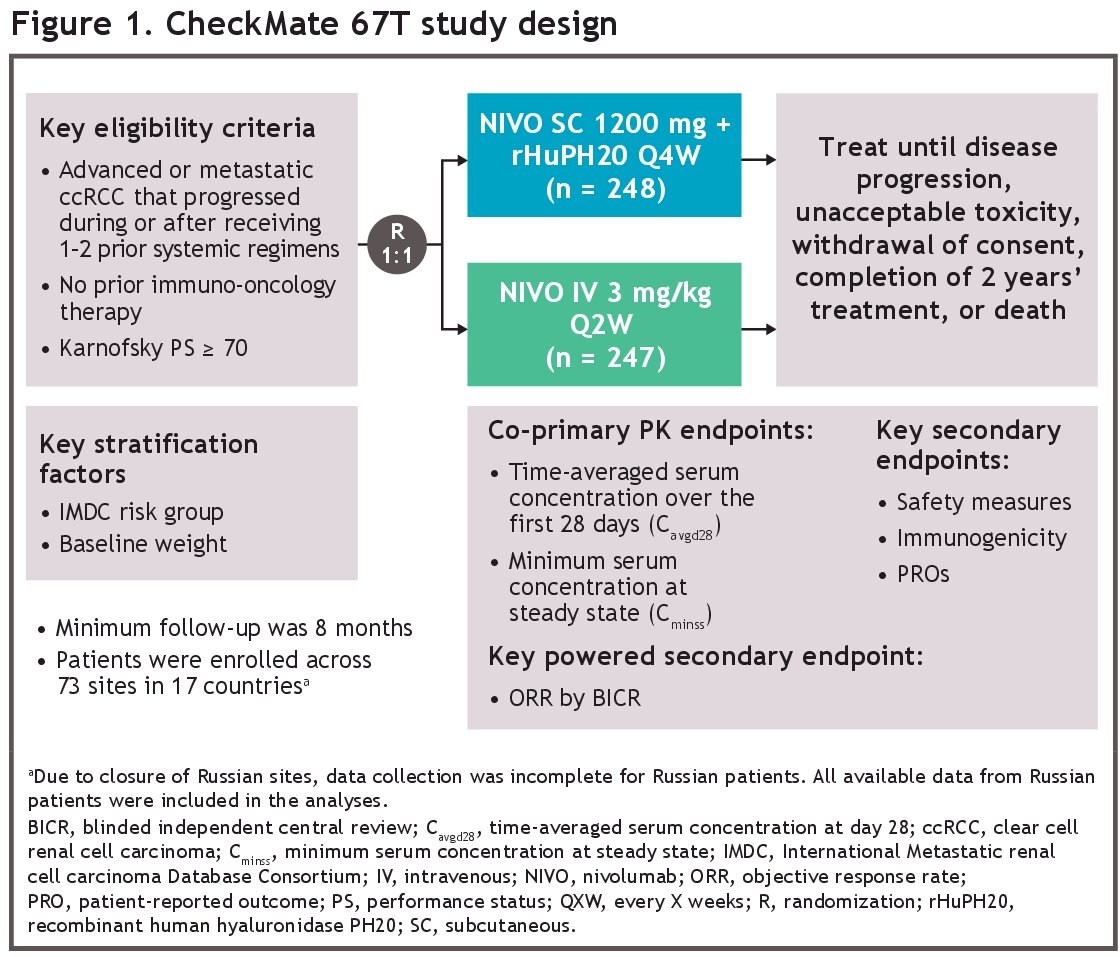
CheckMate 67T (NCT04810078) is a multicenter, randomized, open-label, phase 3 trial evaluating the pharmacokinetics and objective response rate (ORR) noninferiority of subcutaneous nivolumab + rHuPH20 versus intravenous nivolumab in previously treated patients with advanced/metastatic ccRCC:
- The co-primary pharmacokinetic endpoints were met – subcutaneous nivolumab + rHuPH20 demonstrated non-inferiority for exposure (time-averaged serum concentration over the first 28 days and minimum serum concentration at steady state), compared to intravenous nivolumab
- The key powered secondary endpoint was also met – subcutaneous nivolumab + rHuPH20 demonstrated noninferiority for ORR by blinded independent central review, compared to intravenous nivolumab
- The safety profile was consistent between the two arms, and no new safety concerns were identified.
- Nivolumab-related immunogenicity was as expected for subcutaneous administration; data from further immunogenicity assessments did not demonstrate any apparent clinically meaningful impact on pharmacokinetics, efficacy, or safety.
In this report, Dr. Bourlon and colleagues summarized the additional safety analyses and patient-reported outcomes from CheckMate 67T.
In this study, patients were stratified by their International Metastatic RCC Database Consortium (IMDC) risk grouping and baseline weight, and subsequently randomized 1:1 to either:
- Nivolumab 1,200 mg + rHuPH20 20,000 units subcutaneously every 4 weeks (n=248)
- Nivolumab 3 mg/kg intravenously every 2 weeks (n=247)
All-causality adverse events (AEs) were analyzed across the following weight categories: < 50 kg, ≥ 50 kg to < 70 kg, ≥ 70 to < 90 kg, ≥ 90 kg to < 110 kg, and ≥ 110 kg. Data were collected on the onset, management, and resolution of treatment-related select AEs, as well as the proportion, duration, and grade of local site reactions. The effect of nivolumab-specific anti-drug antibodies (ADAs) on local site reactions, hypersensitivity, and infusion-related reaction select AEs was assessed. Patient perception of "bother by treatment side effects" was assessed using the Functional Assessment of Chronic Illness Therapy (FACIT) GP5 item every four weeks, and then during the follow-up period.
A total of 248 and 247 patients in the subcutaneous and intravenous nivolumab arms received treatment, respectively. The median patient age was 64 years (range, 35–93) and 66 years (range, 20–87) years in the two arms, respectively. Approximately one-third of patients were female (33.9% in the subcutaneous arm and 30.8% in the intravenous arm). The median weight was 77 Kg in both arms.
Across different patient weight categories, the overall AE incidence rates in the subcutaneous arm were generally comparable to or lower than those observed in the intravenous arm for most categories across all-causality adverse events (Figure 2).

The incidence of treatment-related select AEs was comparable between the two arms (Table 1).
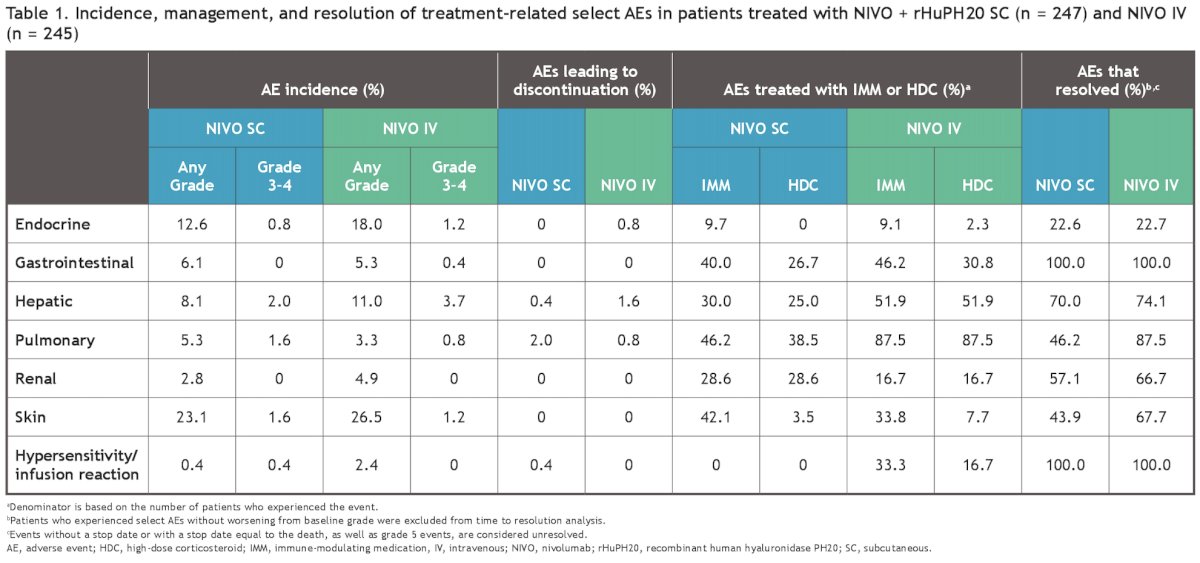
Most of these AEs were manageable using established algorithms and resolved with immune-modulating medications (mainly systemic corticosteroids; Figure 3).
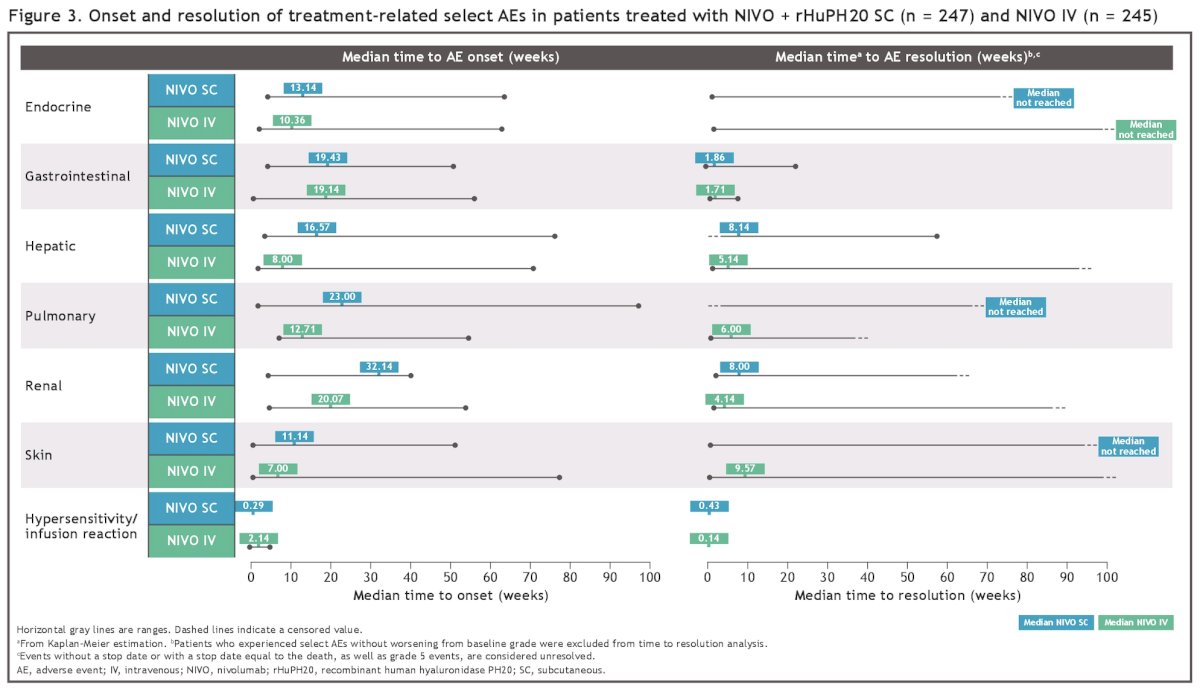
The proportion of patients with a local injection-site reaction was higher in the subcutaneous injection arm (8.1% versus 2%). Local site reactions were mostly grade 1, with a median duration of 2 and 0.01 days in the subcutaneous and intravenous arms, respectively. No grade 3 or higher reactions were reported. Most adverse events are resolved without treatment.
In the patients evaluable for ADA testing, local injection-site reactions were reported in a greater proportion of ADA-positive patients than ADA-negative patients in the subcuteanous arm (15.2% versus 6.5%). In the in intravenous arm, no ADA-positive patients versus 2% of ADA-negative patients reported local infusion-site reactions. All local site reactions were grade 1–2 and resolved without treatment.
No hypersensitivity or infusion/injection-related reaction select AEs were reported in ADA-positive patients in either arm. Hypersensitivity or infusion/injection-related reaction select AEs were reported in 1.3% of ADA-negative patients in the subcutaneous arm and 4% of ADA-negative patients in the intravenous arm.
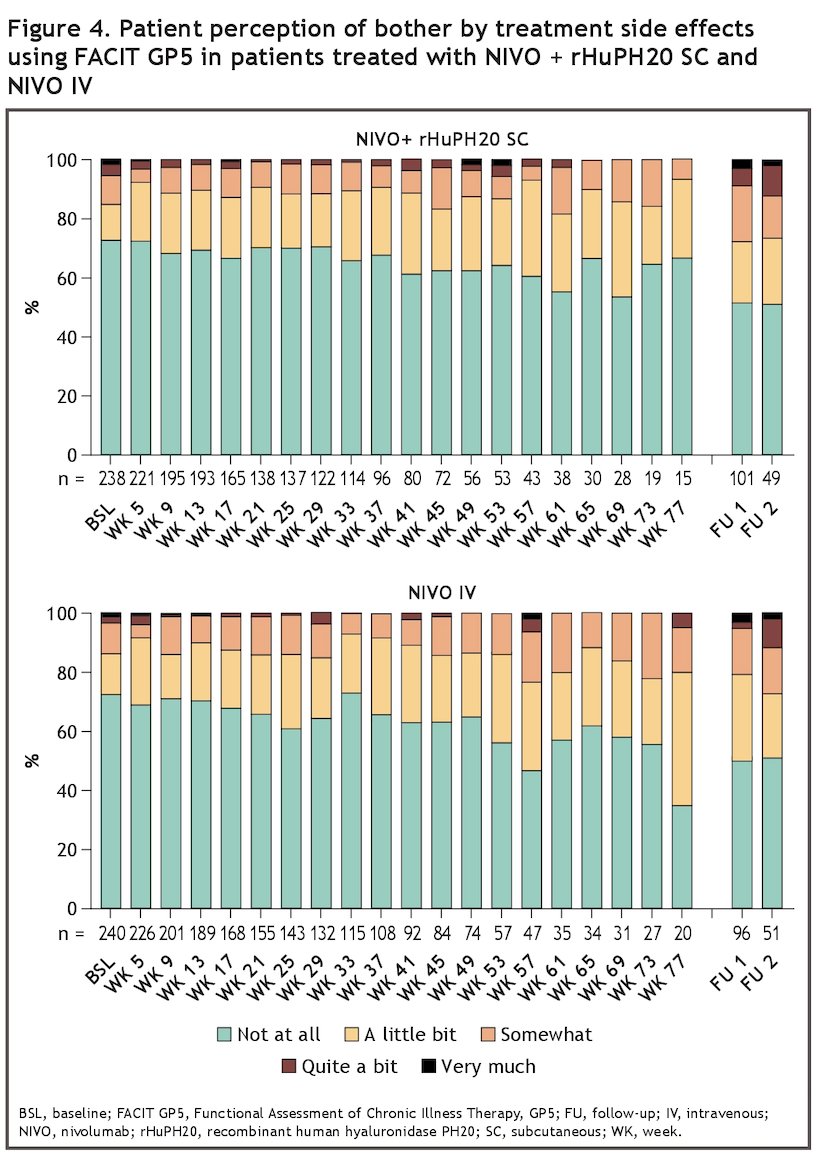
With regards to patient experience of treatment side effects, using the FACIT GP5 item, the proportion of patients bothered by treatment side effects was shown to be low in both treatment arms (Figure 4).
Dr. Bourlon concluded as follows:
- The overall safety profile of subcutaneous nivolumab was consistent with that of intravenous nivolumab, and no new safety concerns were identified
- Additional analyses of safety by weight categories were consistent with the overall safety profile, with incidence rates in the subcutaneous arm being comparable to or lower than those in the intravenous arm for most categories across all-causality adverse events
- The safety profile was manageable based on the already established management guidelines
- There was no apparent clinically meaningful impact of anti-nivolumab antibodies on the safety of the subcutaneous or intravenous arms
- The proportion of patients bothered by treatment side effects was low and generally similar between the two treatment arms
- These data, along with previously presented data from this study, continue to indicate that subcutaneous nivolumab provides clinical equipoise to standard IV dosing, supporting the use of subcutaneous nivolumab as a new option to reduce patient treatment burden and improve healthcare efficiency
Presented by: Maria T. Bourlon, MD, MSc, Urologic Oncology Clinic, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico
Written by: Rashid Sayyid, MD, MSc – Society of Urologic Oncology (SUO) Clinical Fellow at The University of Toronto, @rksayyid on Twitter during the 2024 American Society of Clinical Oncology (ASCO) Annual Meeting, Chicago, IL, Fri, May 31 – Tues, June 4, 2024.