(UroToday.com) The 2024 IBCN annual meeting included a keynote lecture by Dr. Niko Beerenwinkel discussing inferring tumor evolution from single-cell data. Although the majority of work has been done in other cancers (ie. melanoma, breast cancer) and not bladder cancer, the hope, and goal is that this technology and applied techniques will soon be used in bladder cancer.
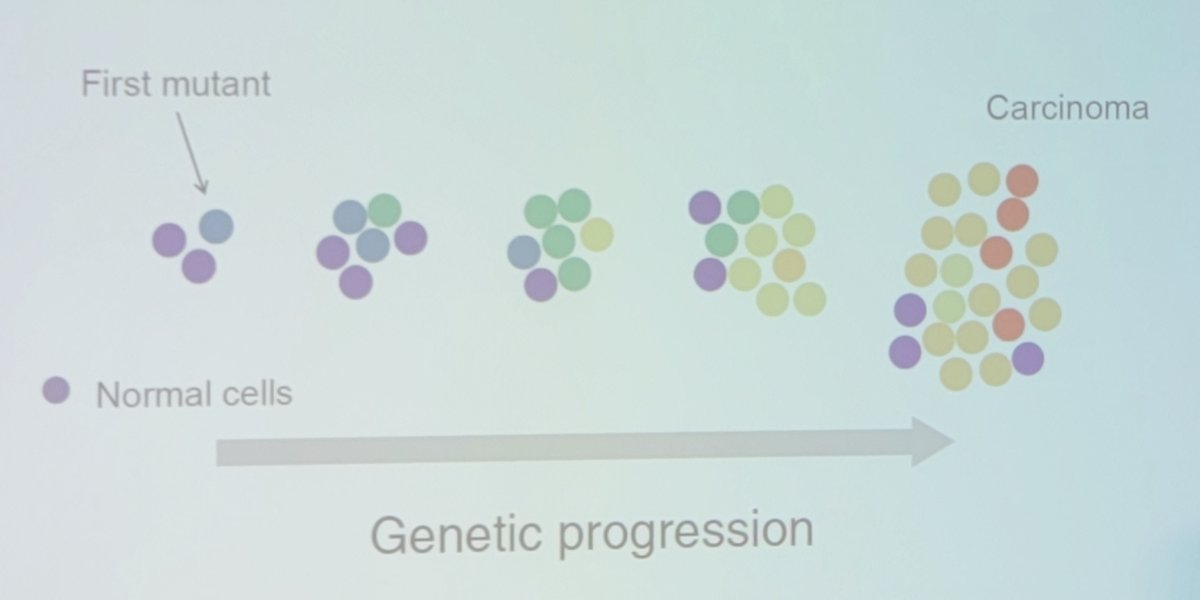
Dr. Beerenwinkel started by noting that cancer is an evolutionary process, likely starting with a first mutant amongst normal cells and subsequently leading to carcinoma:
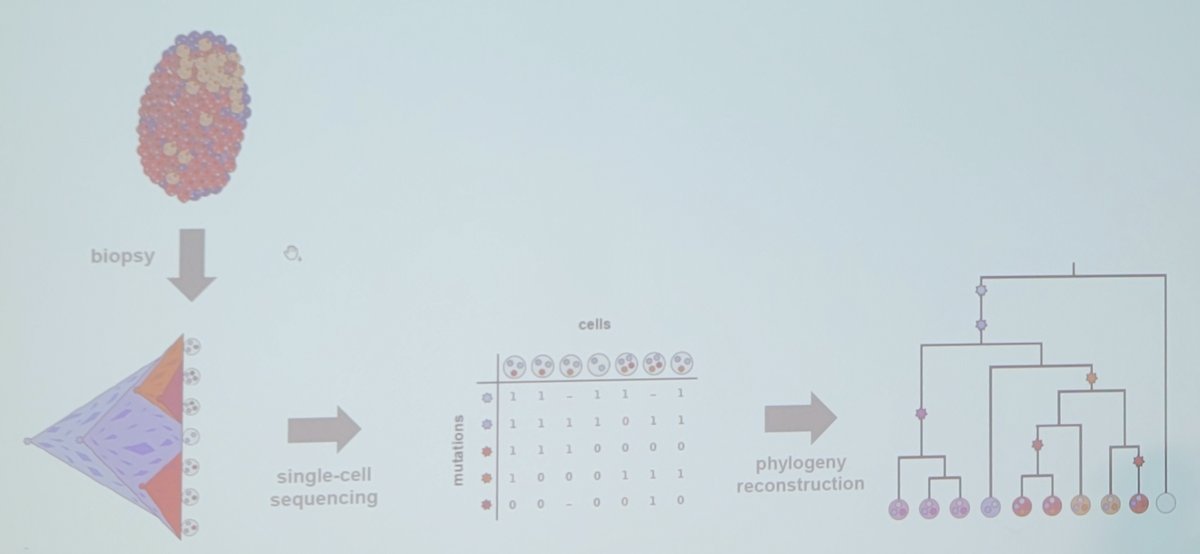
Another way of looking at this concept is tumor phylogeny:

At a high level, the tumor phylogeny reconstruction problem is highlighted by a biopsy, which leads to single-cell sequencing, which leads to phylogeny reconstruction:
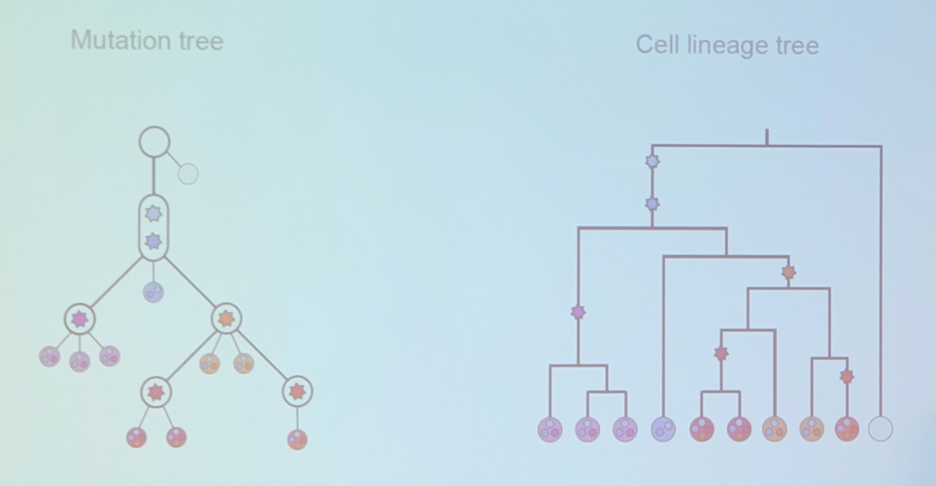
What most of us are likely familiar with is a mutation tree, but the next step in development is a cell lineage tree:
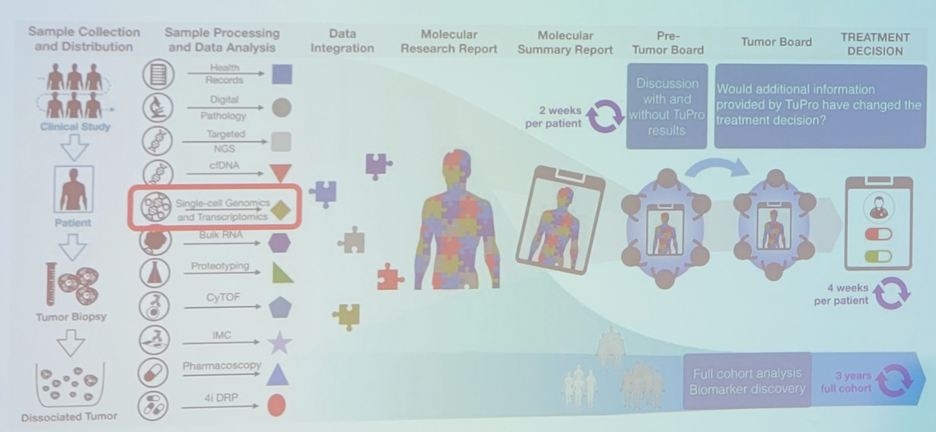
Dr. Beerenwinkel then discussed a large project he is involved with in Switzerland called the Tumor Profiler Study, assessing multiomics tumor profiling for clinical decision support. As part of this project, Dr. Beerenwinkel’s role is in single-cell genomics and transcriptomics:
Dr. Beerenwinkel then discussed single-cell copy number and single nucleotide variant calling and phylogenetic tree reconstruction. Specifically, SCICoNE (single cell copy number calling and event history reconstruction) starts with the data, subsequently modeling, and finally a prediction model:

At a more detailed level is joint single nucleotide variant and copy number variant single-cell tumor phylogenies (panel sequencing data):

Dr. Beerenwinkel then discussed the mapping of single-cell transcriptomes to copy number evolutionary trees (SCATrEx). The question is, does the genomic diversity correlate with transcriptomic diversity?
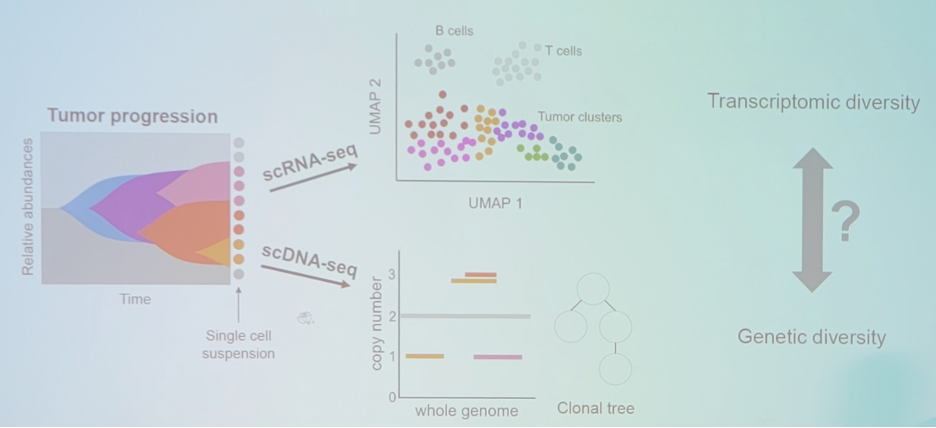
Dr. Beerenwinkel notes that the augmented tree describes tumor development in terms of copy number and gene expression changes:

Importantly, Dr. Beerenwinkel notes that SCATrEx enables integrated copy number variant gene expression analysis, with the following example from triple negative breast cancer:
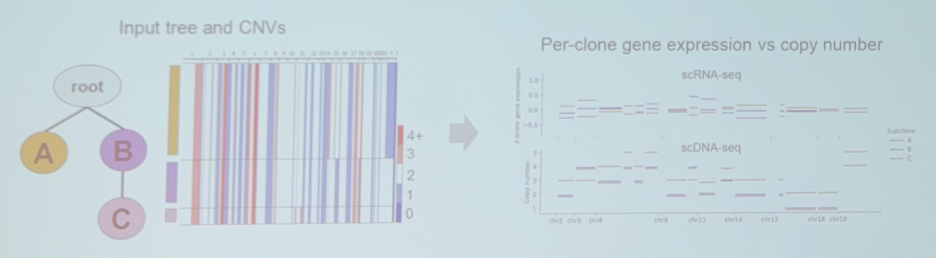
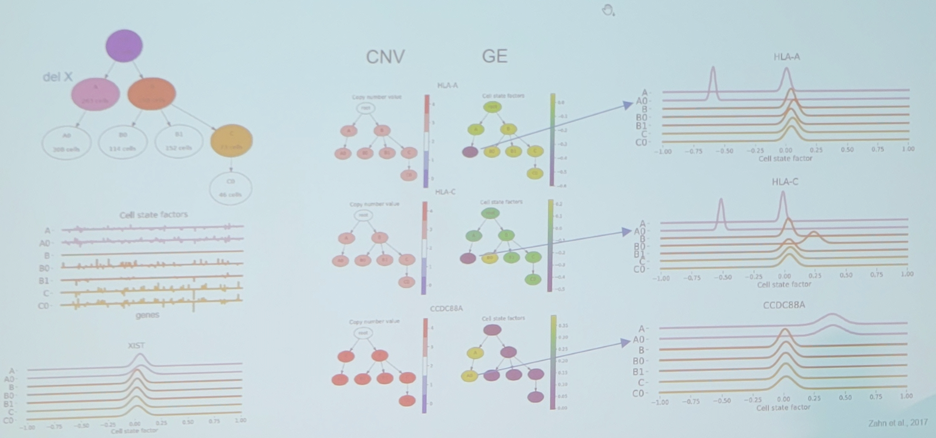
Dr. Beerenwinkel then discussed identifying hierarchical cell states and gene signatures with deep exponential families for single-cell transcriptomics. There are batch effects in single-cell RNA-seq data: (i) no batch considerations – technical and biological batch effects are confounded; (ii) batch overcorrection – technical and biological batch effects are both removed. Thus, can we find a balance between these two? One potential avenue is single-cell deep exponential families (scDEF) with the following overview:
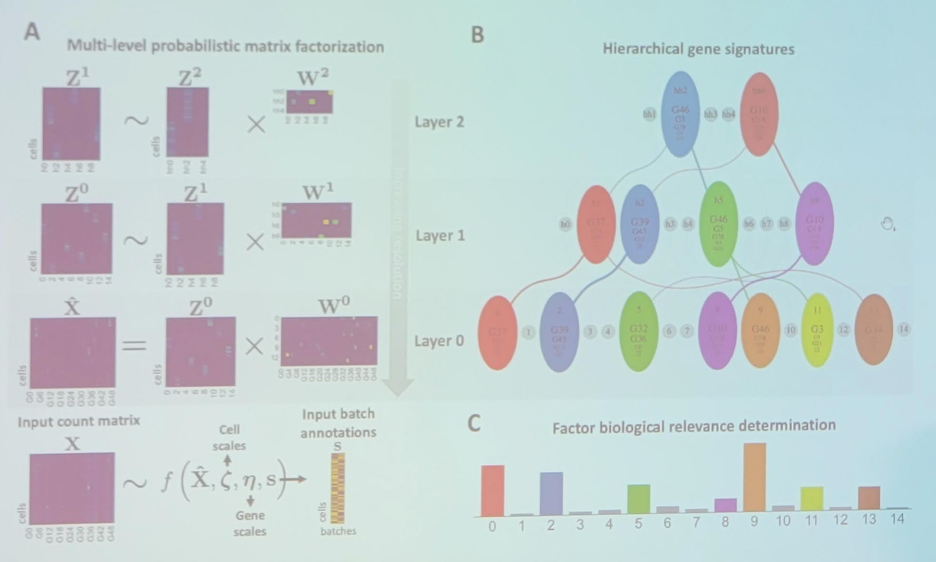
Finally, Dr. Beerenwinkel discussed the single-cell transcriptomic and genomic landscape of metastatic melanoma. Ultimately, this leads to integrated single-cell RNA-seq and single-cell DNA-seq on the TPC melanoma cohort, leading to scDEF factor hierarchy, SCATrEx denoised:

Integrating data reveals varying levels of copy number alteration effects. This includes (i) strong copy number alteration influence, (ii) weak copy number alteration effects, and (iii) combined copy number alterations and transcriptional regulation:
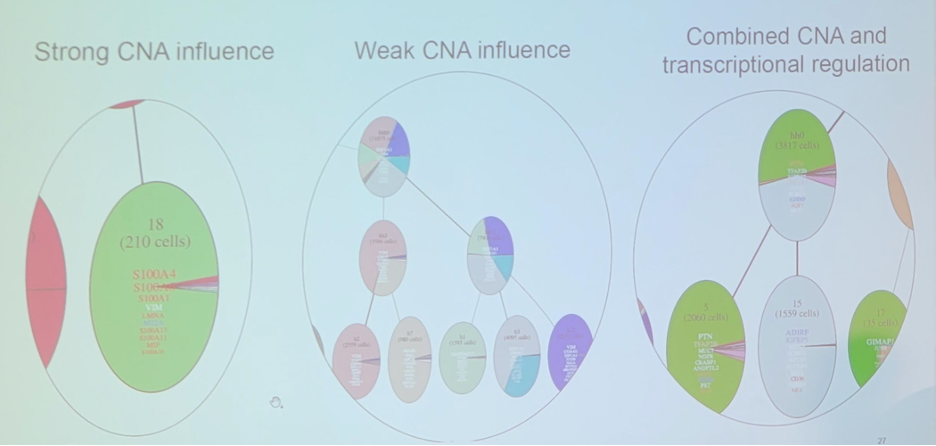
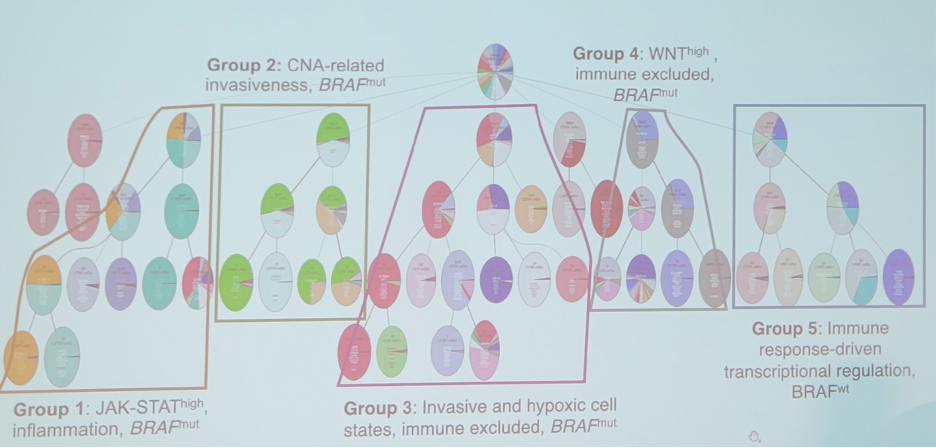
Furthermore, integrated data reveals evolutionary-driven functional subgroups:
- Group 1: JAK-STAThigh, inflammation, BRAPmut
- Group 2: CNA-related invasiveness, BRAPmut
- Group 3: Invasive and hypoxic cell states, immune excluded, BRAPmut
- Group 4: WNThigh, immune excluded, BRAPmut
- Group 5: Immune response driven transcriptional regulation, BRAPwt
Dr. Beerenwinkel concluded his presentation discussing inferring tumor evolution from single-cell data with the following take-home points:
- Single-cell tumor phylogenies:
- Allows reconstruction of the evolutionary history of a tumor
- Serves as the backbone for integrating and denoising single-cell multiomics data
- Can help identify evolutionary biomarkers
- Can help identify patient subgroups with similar tumor evolutionary patterns
Written by: Zachary Klaassen, MD, MSc – Urologic Oncologist, Associate Professor of Urology, Georgia Cancer Center, WellStar MCG Health, @zklaassen_md on Twitter during the 2024 International Bladder Cancer Network (IBCN) Annual Meeting, Bern, Switzerland, Thurs, Sept 19 – Sat, Sept 21, 2024