ORLANDO, FL, USA (UroToday.com) - It is well known that androgens (testosterone and dihydrotestosterone) fuel tumor growth in prostate cancer, and treatment with LHRH agonists or GnRH antagonists that reduce circulating androgen levels are the standard of care in early-stage disease.
Ketoconazole, a synthetic antifungal drug, has been used off-label to treat prostate cancer because of its inhibition of CYP17 lyase. Recently drug developers have targeted CYP17 inhibition to reduce the biosynthesis of androgens and one drug, abiraterone acetate, is approved in combination with low-dose prednisone (P) for treating castration resistant prostate cancer (CRPC).
 CYP17 inhibitors have been developed to treat CRPC because CYP17 has a central role in converting progestogens to androgens. CYP17 has both hydroxylase and lyase catalytic functions. However, selective hydroxylase inhibition causes build-up of progestogens and mineralocorticoids, resulting in secondary mineralocorticoid excess (ME), edema, hypokalemia, and hypertension (treated clinically with prednisone). In this study, four CYP17 inhibitors were analyzed for their effects on CYP17 hydroxylase and lyase activities, and more globally on the steroidogenic pathway.
CYP17 inhibitors have been developed to treat CRPC because CYP17 has a central role in converting progestogens to androgens. CYP17 has both hydroxylase and lyase catalytic functions. However, selective hydroxylase inhibition causes build-up of progestogens and mineralocorticoids, resulting in secondary mineralocorticoid excess (ME), edema, hypokalemia, and hypertension (treated clinically with prednisone). In this study, four CYP17 inhibitors were analyzed for their effects on CYP17 hydroxylase and lyase activities, and more globally on the steroidogenic pathway.
As a result of ME, abiraterone must be co-administered with P, and orteronel is also being dosed with P in Phase III clinical studies. Galeterone is a proprietary oral small-molecule compound that disrupts androgen receptor (AR) signaling via a novel triple mechanism of action: (1) selective inhibition of CYP17 lyase; (2) competitive antagonism of androgen (testosterone, DHT) binding to AR; and (3) degradation of the AR protein itself. Patients treated with galeterone at doses that produced radiographic responses and reductions in PSA have so far not exhibited dose-limiting toxicities or signs of ME and have not required concomitant P (Montgomery, 2012).
Human CYP17 expressed in yeast microsomes was incubated with pregnenolone or 17α- hydroxypregnenolone and products quantitated using LC/MS/MS (Agilent 6410 LC/MS/MS system). Galeterone, abiraterone, orteronel, or ketoconazole were added, and hydroxylase or lyase IC50 values calculated. In a separate experiment, H295R adenocarcinoma cells were incubated with drugs at 1 μM for 24 hours and media was analyzed for steroid production by LC/MS/MS.
All four drugs inhibited CYP17, but there was a significant variation in potency and selectivity for hydroxylase and lyase. Galeterone was the most potent and selective CYP17 lyase inhibitor. Abiraterone most selectively inhibited hydroxylase, while orteronel and ketoconazole were less potent lyase inhibitors than galeterone (see figure below). In the cell-based assay, all drugs inhibited testosterone synthesis≥ 94% at 1 µM. However, at this concentration, evidence of hydroxylase inhibition was supported by significant elevation of progesterone (abiraterone increased 293-fold), mineralocorticoids (orteronel increased DOC 74-fold), or reductions in cortisol (abiraterone, orteronel, ketoconazole reduced by 91%, 70% and 94%, respectively). In contrast, galeterone produced minimal changes in cortisol (decreased 14%) and other intermediate precursors.
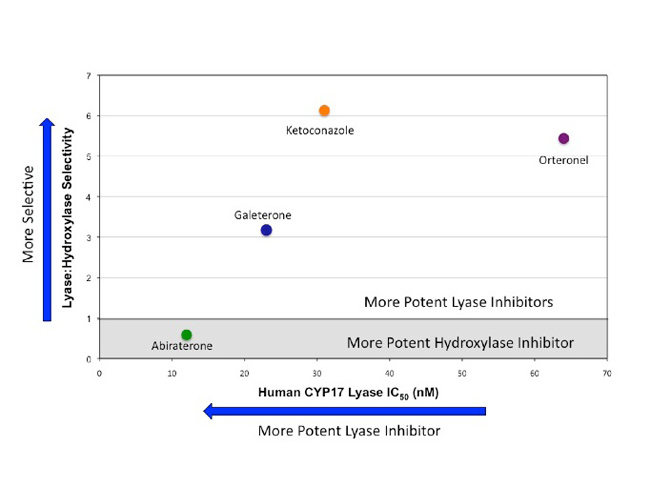
To summarize the properties of the four drugs evaluated in this study:
- Galeterone:
- was the most potent CYP17 inhibitor that was also selective for CYP17 lyase
- caused only moderate elevations of pregnenolone, deoxycorticosterone and corticosterone
- had the smallest effect on the levels of CYP17A1 hydroxylase products, especially cortisol
These experimental data may explain the Phase I clinical experience with galeterone where no ME was observed or P required.
- Abiraterone:
- was the most potent CYP17 inhibitor, but was more selective for CYP17 hydroxylase than lyase
- reduced cortisol and elevated pregnenolone, progesterone, and to a lesser extent deoxycorticosterone and corticosterone
These changes are consistent with the ME observed clinically.
- Orteronel:
- was a less potent, selective CYP17 lyase inhibitor
- elevated CYP17A1 hydroxylase precursors (especially deoxycorticosterone and corticosterone) and reduced CYP17A1 hydroxylase products, including cortisol
The effects are consistent with inhibition of CYP17A1 hydroxylase which could cause ME.
- Ketoconazole:
- was a less potent, selective CYP17 lyase inhibitor
- exhibited dose-dependent inhibition of CYP17A1 hydroxylase and lyase, but also caused off-target effects in the pathway.
Non-specific effects of ketoconazole on other CYP450 enzymes limit its clinical use due to toxicity.
To conclude, clinical experience with galeterone has shown no signs of ME, and it has not required concomitant P use to date. This study demonstrated that the differential clinical profile of galeterone is ascribed to its highly selective and potent CYP17 lyase inhibition, while minimally impacting CYP17 hydroxylase function.
Presented by Douglas Burtner Jacoby and Martin Williams at the 2013 Genitourinary Cancers Symposium - February 14 - 16, 2013 - Rosen Shingle Creek - Orlando, Florida USA
Tokai Pharmaceuticals, Cambridge, MA
Prepared by Anna Forsberg, medical writer for UroToday.com
View Full 2013 GU Cancers Symposium Coverage