The majority of ccRCC patients carry inactivation of the von Hippel-Lindau (VHL) gene leading to genetic stabilisation of hypoxia-inducible factor (HIF) transcription factor alpha. The HIF pathway drives tumor development and progression in the VHL–inactivated ccRCC. HIF transcriptionally targets over 100 genes,1 and the loss of VHL function induces constitutive HIF-1α/2α expression that markedly upregulated their target genes, including vascular endothelial growth factor (VEGF) and erythropoietin (EPO). Consequently, ccRCC are hyper vascularized tumors with frequent mutations in chromosome 3p, that affect an array of chromatin-remodeling genes, including Polybromo 1 (PBRM1), SET Domain Containing 2 (SETD2), and BRCA1 Associated Protein 1 (BAP1).2,3
Tyrosine kinase inhibitors (TKI) primarily targeting VEGF receptors such as sunitinib are the first-line therapy for the treatment of metastatic ccRCC during the last decade.4 Sunitinib inhibits angiogenesis by blocking VEGFRs. In addition to its anti-angiogenic effects on endothelial cells, sunitinib directly targets tumor cells to inhibit their proliferation, migration, and survival.5,6 ccRCC patients receiving sunitinib ineluctably develop resistance through a genetic adaptation of tumor cells leading to their survival in the presence of the drug.
Our study7 demonstrated that sunitinib-resistant ccRCC cells exhibit higher Polo-Like Kinase 1 (Plk1) expression. This result suggests that Plk1 induction is part of a genetic program associated with resistance to sunitinib.
Polo-Like Kinase 1 (Plk1) is a serine/threonine kinase which acts during cell cycle progression.8 Plk1 inhibits p53, and p53 represses the Plk1 promoter.9 High Plk1 expression correlates with an advanced disease stage, histological grades, metastatic potentials, and short-term survival in various tumors.10 The Plk1 inhibitor volasertib inhibits the proliferation of a variety of carcinoma cell lines and induces tumor regression in several experimental tumor models.11
Before our study, the molecular mechanisms linking Plk1 expression and tumor hypoxia were unknown.
In the context of ccRCC, the absence of VHL (mutation, deletion, methylation) leads to HIF chronic stabilization and to the up-regulation of the transcription of target genes independently of the oxygen concentration.
We showed that hypoxia-dependent up-regulation of Plk1 depends on increased transcription dependent on HIF-2 but not HIF1, and on the mutation of SETD2 in human ccRCC. HIF-1α is considered as a tumor suppressor whereas HIF-2α is considered as an oncogene in ccRCC.12
Metastatic ccRCC patients relapse despite inhibition of angiogenesis (with VEGFR-TKI like sunitinib) and immune checkpoint inhibitors. Predictive markers of efficacy of the current treatments and new therapeutic targets are urgently needed for patients in therapeutic impasses. Thus, Plk1 blockade represents an attractive and alternative therapeutic solution for treating sunitinib-resistant ccRCC patients. Indeed, we provided compelling evidence showing that sunitinib-resistant ccRCC cells are highly sensitive to Plk1 inhibition. Targeting Plk1 inhibited tumor and endothelial cell proliferation in mice models. Therefore, the therapeutic efficacy of Plk1 inhibitors also relies on the inhibition of angiogenesis, a key phenomenon in ccRCC. We also showed that aggressive ccRCC cells metastasized in zebrafish’ tails without genetic modification beforehand and Plk1 inhibition prevented this metastatic spreading. This exciting finding warrants clinical validation.
Moreover, we demonstrated that Plk1 is a driver of tumor growth orchestrated by the HIF-2 oncogenic pathway in ccRCC. Indeed, Plk1 is linked to shorter survival in both non-metastatic and metastatic patients. It is a prognostic factor independent of the clinical prognostic IMDC score. Hence, a biological marker independent of clinical parameters provides an added value to the management of patients.
The new gold standards for metastatic ccRCC patients in the first-line are TKI, immunotherapy (anti-PD1 + anti-CTLA413), or a combination of both therapies14,15
Based on gene expression, methylation status, mutation profile, cytogenetic anomalies, and immune cell infiltration, 4 subtypes of ccRCC patients (ccrcc1–4) have been determined.16,17 These markers have prognostic and predictive values for guiding TKI-based therapy. The ccrcc2&3 subtypes possess a good prognostic value of progression-free (PFS) and overall survival (OS) and favorable for TKI therapy, whereas the ccrcc1&4 subtypes have the opposite prognostic values with poor prognosis and TKI responses. The ccrcc2-tumors often express proangiogenic genes and ccrcc3-tumors resemble healthy kidney tissue according to gene expression profiling. Ccrcc4-tumors exhibit an immune-inflamed phenotype, but an exhausted immune system. The ccrcc1-tumors belong to an immune-cold phenotype almost without lymphocyte infiltration.16,17 Therefore, the ccrcc2&3-tumors are eligible for TKI therapy and the ccrcc4-tumors are potential good responders to immunotherapy (BIONIKK clinical trial, NCT0296090618). In contrast, ccrcc1-tumors fail to respond to either therapy.
Tumors of the ccrcc1 subtype strongly express Plk1. High Plk1 mRNA levels correlated with a poor response to immunotherapy.19 Therefore, Plk1 inhibition represents a relevant strategy for these patients. The following nomogram appears decisional for the therapeutic strategy for patients of the different subgroups:
- ccrcc2&3 subtypes (low PDL1 and Plk1 expression) are eligible for TKI,
- ccrcc4 subtype (high PDL1 and Plk1 expression) are eligible for immunotherapy,
- ccrcc1 subtype (low PDL1 expression but strong Plk1 expression) are eligible for treatment with Plk1 inhibitors.
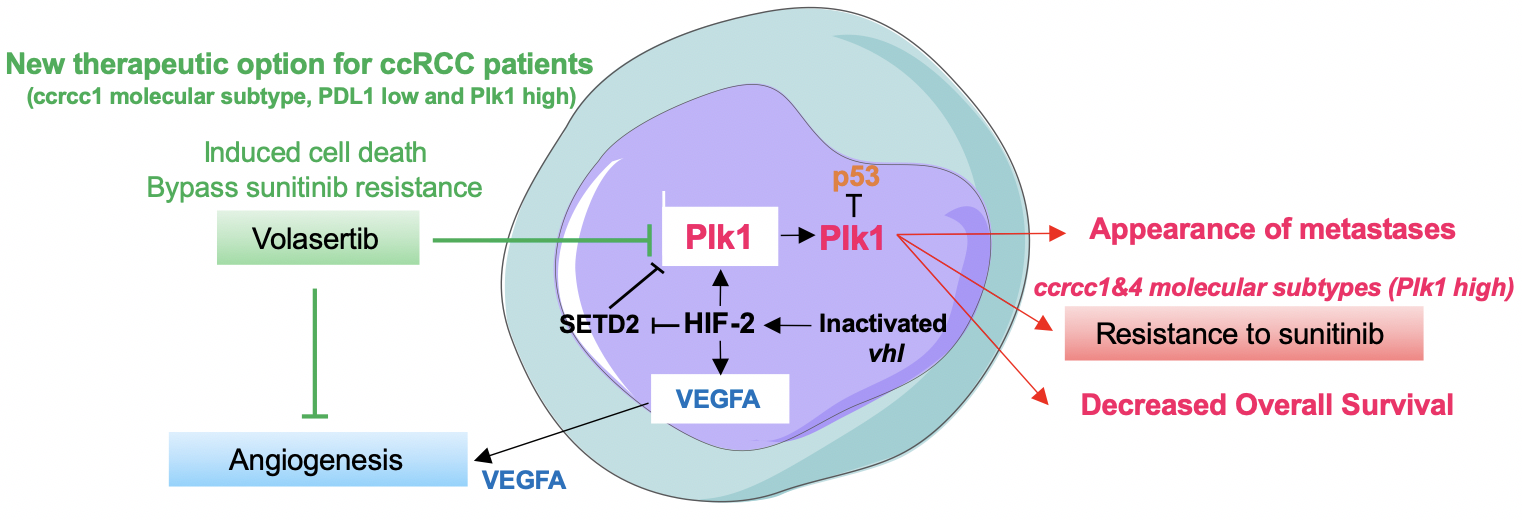
Figure 1. The summary diagram of our study describing the link between HIF-2-dependent driving of Plk1 gene transcription through a SETD2-dependent mechanism. In addition to its tumor-promoting role, Plk1 drives resistance to TKI and appears as a key target for a subgroup of metastatic ccRCC patients in therapeutic impasses 7
In brief, our study deciphered the phenomenon linking a physical driver of tumor aggressiveness (hypoxia, HIF-2) to a biological determinant of tumor cell proliferation and angiogenesis (Plk1). The link between the two actors is HIF-2, which drives Plk1 gene transcription. In addition to its tumor-promoting role, Plk1 drives resistance to TKI and appears as a key target for a subgroup of metastatic ccRCC patients in therapeutic impasses (Figure 1).
Written by: Maeva Dufies1,2 and Gilles Pagès1,2,3
- Centre Scientifique de Monaco, Biomedical Department, 8 quai Antoine Premier, 98 000 Monaco, Principality of Monaco
- LIAROPSE, Laboratoire International Associé Université Côte d’Azur - Centre Scientifique de Monaco
- University Côte d’Azur, Institute for Research on Cancer and Aging of Nice (IRCAN), CNRS UMR 7284; INSERM U1081, Centre Antoine Lacassagne, Nice, France
References:
- Manalo, D. J. et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105, 659–669 (2005).
- Hsieh, J. J. et al. Chromosome 3p Loss–Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. JCO 36, 3533–3539 (2018).
- Ricketts, C. J. & Linehan, W. M. Insights into Epigenetic Remodeling in VHL-Deficient Clear Cell Renal Cell Carcinoma. Cancer Discov 7, 1221–1223 (2017).
- Eisen, T. et al. Targeted Therapies for Renal Cell Carcinoma: Review of Adverse Event Management Strategies. JNCI Journal of the National Cancer Institute 104, 93–113 (2012).
- Giuliano, S. et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 11, 1891–1904 (2015).
- Dufies, M. et al. Sunitinib Stimulates Expression of VEGFC by Tumor Cells and Promotes Lymphangiogenesis in Clear Cell Renal Cell Carcinomas. Cancer Res. 77, 1212–1226 (2017).
- Dufies, M. et al. Plk1, upregulated by HIF-2, mediates metastasis and drug resistance of clear cell renal cell carcinoma. Commun Biol 4, 166 (2021).
- Liu, Z., Sun, Q. & Wang, X. PLK1, A Potential Target for Cancer Therapy. Translational Oncology 10, 22–32 (2017).
- Louwen, F. & Yuan, J. Battle of the eternal rivals: restoring functional p53 and inhibiting Polo-like kinase 1 as cancer therapy. Oncotarget 4, 958–971 (2013).
- Weng Ng, W. T., Shin, J.-S., Roberts, T. L., Wang, B. & Lee, C. S. Molecular interactions of polo-like kinase 1 in human cancers. J Clin Pathol 69, 557–562 (2016).
- Gutteridge, R. E. A., Ndiaye, M. A., Liu, X. & Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther 15, 1427–1435 (2016).
- Biswas, S. et al. Effects of HIF-1alpha and HIF2alpha on Growth and Metabolism of Clear-Cell Renal Cell Carcinoma 786-0 Xenografts. J Oncol 2010, 757908 (2010).
- Motzer, R. J. et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 378, 1277–1290 (2018).
- Motzer, R. J. et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 380, 1103–1115 (2019).
- Rini, B. I. et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 380, 1116–1127 (2019).
- Beuselinck, B. et al. Molecular Subtypes of Clear Cell Renal Cell Carcinoma Are Associated with Sunitinib Response in the Metastatic Setting. Clin Cancer Res 21, 1329–1339 (2015).
- Verbiest, A. et al. Clear-cell Renal Cell Carcinoma: Molecular Characterization of IMDC Risk Groups and Sarcomatoid Tumors. Clinical Genitourinary Cancer 17, e981–e994 (2019).
- Epaillard, N. et al. BIONIKK: A phase 2 biomarker driven trial with nivolumab and ipilimumab or VEGFR tyrosine kinase inhibitor (TKI) in naïve metastatic kidney cancer. Bulletin du Cancer 107, eS22–eS27 (2020).
- Li, M., Liu, Z. & Wang, X. Exploration of the Combination of PLK1 Inhibition with Immunotherapy in Cancer Treatment. Journal of Oncology 2018, 1–13 (2018).
Read the Abstract