Introduction
Given the promising results that Poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors have demonstrated for the treatment of metastatic castrate-resistant prostate cancer (mCRPC) patients who had progressed following prior androgen receptor pathway inhibitor (ARPI) and/or taxane-based chemotherapy, there has been increased interest in ‘moving up’ these agents along the disease spectrum, as well as combining them with other agents that may have a synergistic mechanism of action.In 2023, three PARP inhibitor/ARPI combinations received US Food and Drug Administration (FDA) approval for the treatment of mCRPC patients in the 1st line setting:
- Olaparib plus abiraterone for BRCA1/2-mutated patients1
- Niraparib plus abiraterone for BRCA1/2-mutated patients2
- Talazoparib plus enzalutamide for homologous recombination repair (HRR)-mutated patients3
PARP Inhibitors + Androgen Receptor Pathway Inhibitors
Preclinical models have suggested synergistic mechanisms of action for PARP inhibitors and ARPIs. PARP inhibitors upregulate androgen receptor signaling, enhancing ARPI activity. Conversely, ARPIs inhibit the transcription of some HRR genes, inducing an HRR deficiency-like state, thus potentiating PARP inhibitor activity.4-6 These mechanisms of action suggest that the PARP inhibitors/ARPI combination may have clinical efficacy, irrespective of HRR mutational status.PROpel: Olaparib + Abiraterone Acetate/Prednisone
PROpel is a global, randomized, double-blind phase 3 trial of abiraterone and olaparib versus abiraterone and placebo in patients with mCRPC treated in the first-line setting. Patients in PROpel were enrolled irrespective of HRR mutational status, which was ascertained via circulating tumor DNA (ctDNA) or tissue testing. Patients were randomized 1:1 to receive abiraterone (1000 mg once daily) plus prednisone/prednisolone with either full dose olaparib (300 mg twice daily) or placebo. Prior abiraterone use was not permitted, but other ARPI use was permitted if discontinued ≥12 months prior to study enrollment. The primary endpoint was imaging-based progression-free survival, by investigator assessment. The trial design for PROpel is as follows: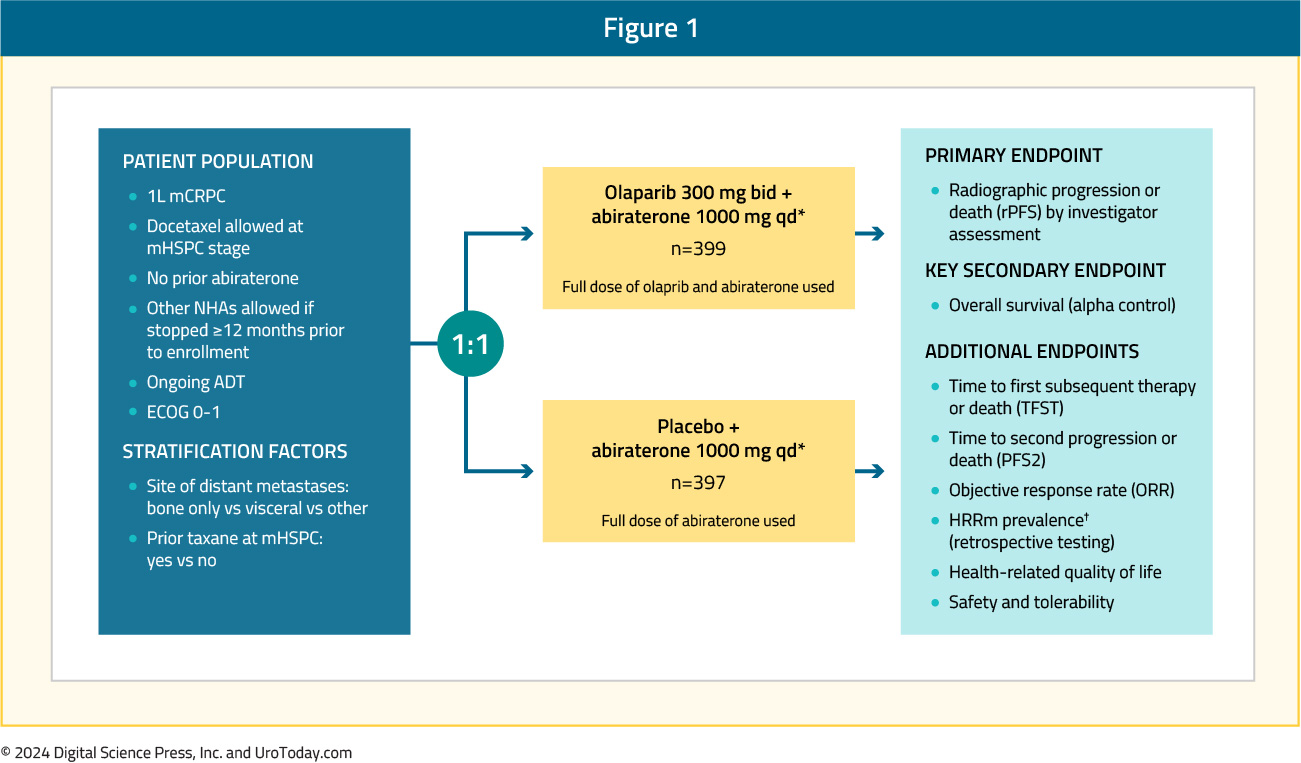
PROpel randomized 399 and 397 patients to the PARP inhibitor + ARPI and placebo + ARPI arms, respectively. The initial results were published in the NEJM Evidence in 2022.7 The median patient age was 70 years, with a median PSA of 17 – 18 ng/ml, and approximately 25% had received prior docetaxel. The most frequent site of distant metastases was the bone (85- 88%). Overall, 29% of patients harbored HRR mutations, with BRCA1/2 mutations present in 10.7% of patients. At a median follow-up of 19.4 months, the combination of olaparib + abiraterone significantly improved median investigator-assessed rPFS from 16.6 to 24.8 months (HR: 0.66, 95% CI: 0.54 – 0.81, p<0.001). Consistent results were observed when rPFS was assessed via blinded-independent central review (BICR):

rPFS benefits were observed irrespective of HRR mutational status, although, as expected, the magnitude of the effect was higher in the HRR mutation patients (HR: 0.50, 95% CI: 0.34 – 0.73) versus non-HRR mutation patients (HR: 0.76, 95% CI: 0.60 – 0.97). Additionally, on subgroup analysis, rPFS benefits were observed irrespective of prior docetaxel exposure and metastatic site.
The final overall survival (OS) data at a median follow-up of 36.6 months were published in The Lancet Oncology in 2023.8 This demonstrated a trend towards an OS benefit in favor of combination olaparib + abiraterone in the overall intention-to-treat (ITT) population (data maturity: 47.9%; HR 0.81, 95% CI: 0.67 – 1.00, p=0.054), with median OS of 42.1 and 34.7 months, respectively:

Subgroup analysis demonstrated non-significant OS benefits irrespective of HRR mutational status, with a larger magnitude of effect observed in the HRR mutation subgroup (HR: 0.66 versus 0.89 for non-HRR mutations). Although the number of BRCA-mutated patients in this cohort was limited (n=85), there appears to be a clinically meaningful OS benefit in this subgroup (HR: 0.29, 95% CI: 0.14 – 0.56):

The percentage of patients with any treatment-emergent adverse event (AE) or death was similar across both arms (97% and 95%). A higher proportion of patients receiving abiraterone + olaparib (47%) had grade ≥3 AEs, compared to those receiving abiraterone plus placebo (38%). The most common AE was anemia, occurring in 46% of patients receiving olaparib + abiraterone, compared to 16% of patients receiving placebo + abiraterone, with grade ≥3 anemia occurring in 15% and 3% of patients, respectively:

Based on these results, the FDA approved the use of olaparib + abiraterone and prednisone (or prednisolone) for adult patients with deleterious or suspected deleterious BRCA-mutated mCRPC.1
MAGNITUDE: Niraparib + Abiraterone Acetate/Prednisone
MAGNITUDE is a phase III, randomized, double-blind, placebo-controlled, multicenter trial that evaluated the combination of niraparib plus abiraterone acetate + prednisone in mCRPC patients receiving first-line treatment. In contrast to PROpel and TALAPRO-2, this trial included biomarker pre-screened cohorts, meaning that all patients had HRR mutational status determined prior to study enrollment. HRR mutational status was determined using tissue and/or blood samples. Patients with a gene alteration (ATM, BRCA1/2, BRIP1, CDK12, CHEK2, FANCA, HDAC2, or PALB2) detected by ≥1 assay were assigned to the HRR+ cohort, whereas those with both assays negative were included in the HRR- cohort. Patients in the HRR+ and HRR- cohorts underwent 1:1 randomization to receive either reduced-dose niraparib 200 mg once daily (usual dose: 400 mg) and abiraterone acetate 1,000 mg once daily plus prednisone 5 mg twice daily or placebo + abiraterone + prednisone. Given the lower likelihood of clinical benefit with combination treatment in the HRR- cohort, a futility analysis was preplanned when approximately 200 patients had been enrolled and approximately 125 composite end-point events (the first of either PSA progression, radiographic progression, or death) had occurred. For the HRR+ cohort, primary and secondary endpoints were tested using a pre-specified strategy. The primary rPFS endpoint was powered for and tested first in the BRCA1/2 subgroup and if statistical significance was reached, the remainder of the HRR+ patients would be analyzed. Approximately 50% of HRR+ patients were specified to be BRCA1/2 positive. The trial design for MAGNITUDE is as follows:
Results of the futility analysis in the HRR- cohort (n=233) demonstrated no benefit for combination niraparib + abiraterone versus placebo + abiraterone. The hazard ratio for the composite endpoint was 1.09, with futility defined as a hazard ratio greater than 1.0. As anticipated, additional grade ≥3 AEs occurred with addition of niraparib. Given the increased toxicity with no observed oncologic efficacy, the independent data monitoring committee recommended suspending enrollment in the HRR- cohort.9
The HRR+ cohort included 423 patients, with 212 and 211 randomized to combination niraparib + abiraterone and placebo + abiraterone, respectively. In this cohort, 3% of patients had prior exposure to an ARPI and 23% had received <4 months of prior abiraterone for mCRPC. Median age was 69 years in both arms, and median PSA was higher in the experimental arm (21.4 versus 17.4 ng/ml). Moreover, the median follow-up in the HRR+ cohort was 18.6 months. In the BRCA1/2 subgroup, median rPFS, assessed via BICR, was significantly prolonged with combination niraparib + abiraterone (16.6 versus 10.9 months; HR: 0.53, 95% CI: 0.36 – 0.79, p=0.001). A similar rPFS benefit with combination niraparib + abiraterone was next observed in the HRR+ cohort (16.5 versus 13.7 months; HR: 0.73, 95% CI: 0.56 – 0.96, p=0.022):

The second interim analysis of MAGNITUDE was recently published in the Annals of Oncology. At a median follow-up of 24.8 months, niraparib plus abiraterone significantly prolonged rPFS (19.5 versus 10.9 months; HR: 0.55, 95% CI: 0.39 – 0.78), consistent with the first prespecified interim analysis. rPFS was also prolonged in the overall HRR+ population (HR: 0.76, 95% CI: 0.60 – 0.97).

OS data remained immature of the time of the second interim analysis (72.8% of deaths required for the final OS analysis had been observed). To date, there is no evidence of a significant OS benefit with combination niraparib + abiraterone in the BRCA1/2 subgroup (median 29.3 versus 28.6 months; HR: 0.88, 95% CI: 0.58 – 1.34, p=0.55).

In the HRR+ cohort, a similar incidence of any-grade treatment emergent AEs was observed in both arms (99.5% versus 96.2%). The most common (≥30%) AEs for niraparib plus abiraterone versus placebo plus abiraterone, regardless of causality, were anemia (50% versus 23%), hypertension (33% versus 22%), and constipation (33% versus 16%). Transfusion support for anemia was required by 27.4% of patients in the niraparib plus abiraterone group versus 5.2% of patients in the placebo plus abiraterone group. Grade ≥3 AEs were observed in 72% of patients in the niraparib plus abiraterone group and 49% of patients in the placebo plus abiraterone group, of which the most common (≥10%) were anemia (30% versus 9%) and hypertension (16% versus 12%).10
Based on these results, the FDA approved the combination of niraparib and abiraterone acetate with prednisone for adult patients with deleterious or suspected deleterious BRCA-mutated mCRPC.2
TALAPRO-2
TALAPRO-2 is a phase III randomized, double-blind, placebo-controlled trial that randomized mCRPC patients 1:1 to talazoparib 0.5 mg once daily (reduced dose from standard of 1 mg) plus enzalutamide 160 mg once daily versus placebo + enzalutamide. Prior use of docetaxel and abiraterone in the mHSPC, but not in the mCRPC setting, was permitted. No prior use of an androgen receptor pathway inhibitor was permitted. Similar to PROpel, this was a biomarker unselected cohort of ‘all comers’. The primary endpoint was rPFS, assessed via blinded independent central review, and OS was a key secondary endpoint. The trial design for TALAPRO-2 is as follows: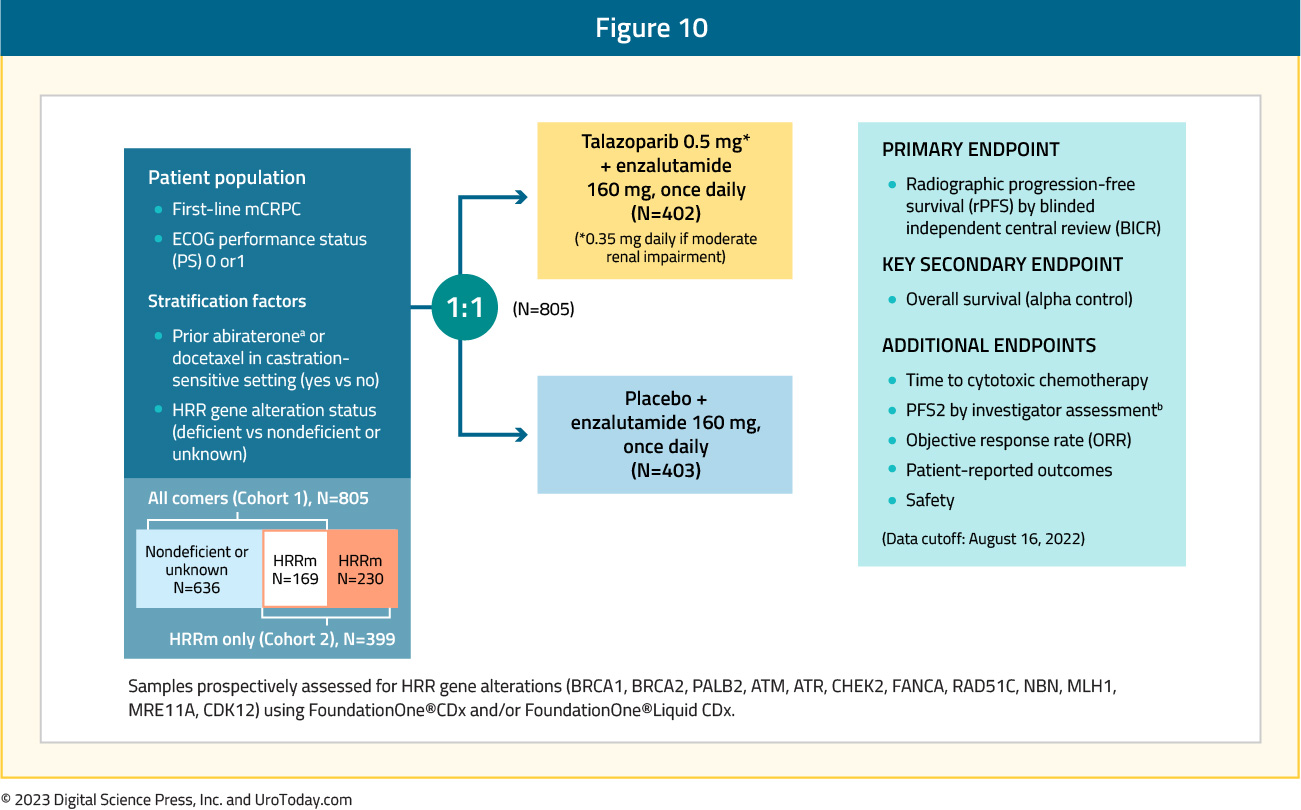
The median age was 71 years, with median serum PSA slightly higher in the intervention arm (18.2 versus 16.2 ng/ml), and 21% of patients in each arm had HRR deficient status. The combination of talazoparib + enzalutamide was associated with a 37% decrease in the rPFS hazard, compared to placebo + enzalutamide (median: not reached versus 22 months; HR: 0.63, 95% CI: 0.51 – 0.78, p<0.001):

Similar to PROpel, rPFS benefits were observed in both the HRR mutated and non-mutated subgroups. As expected, the magnitude of effect was larger in the HRR mutated (median rPFS: 28 versus 16 months; HR: 0.46, 95% CI: 0.30 – 0.70, p<0.001) versus non-mutated subgroup (HR: 0.66, 95% CI: 0.49 – 0.91, p=0.009).

OS data remains immature, with a trend towards an OS improvement in the overall cohort (HR: 0.89, 95% CI: 0.69 – 1.14, p=0.35). Grade ≥3 AEs occurred more commonly in the intervention arm (75% versus 45%), with grade ≥3 anemia observed in 46.5% and 4.2% of patients, respectively. The onset of grade 3-4 anemia occurred at a median time of 3.3 months. The most common treatment emergent AEs resulting in talazoparib dose reductions were anemia (43.2%), neutropenia (15.1%), and thrombocytopenia (5.5%).11
These results served as the basis for the FDA approval of talazoparib plus enzalutamide for HRR gene-mutated mCRPC patients.3
More recently, updated results from the HRR-deficient population were published in Nature Medicine.12 There continued to be a strong, consistent rPFS benefit in favor of talazoparib + enzalutamide (HR: 0.45, 95% CI: 0.33 – 0.61, p< 0.0001):
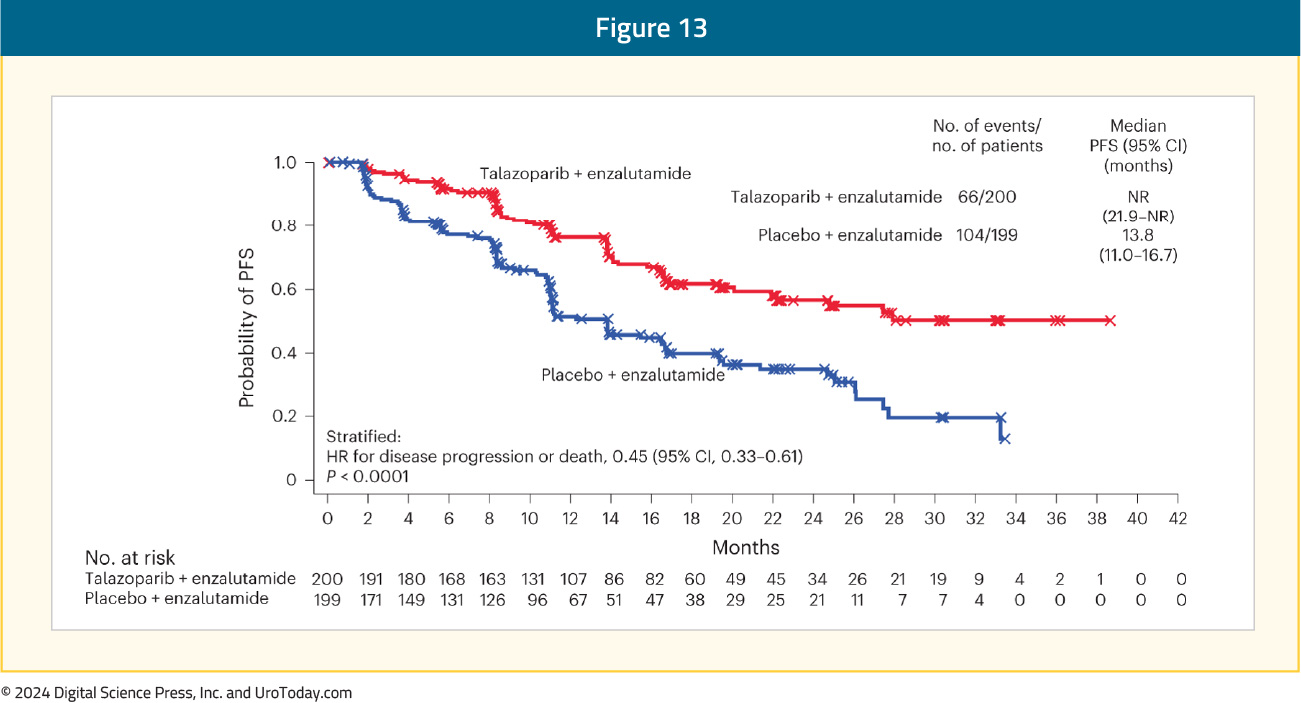
Additionally, there was a non-significant OS benefit in favor of talazoparib plus enzalutamide in the HRR-mutated population (median not reached versus 33.7 months; HR: 0.59, 95% CI: 0.46 – 1.03, p=0.07).
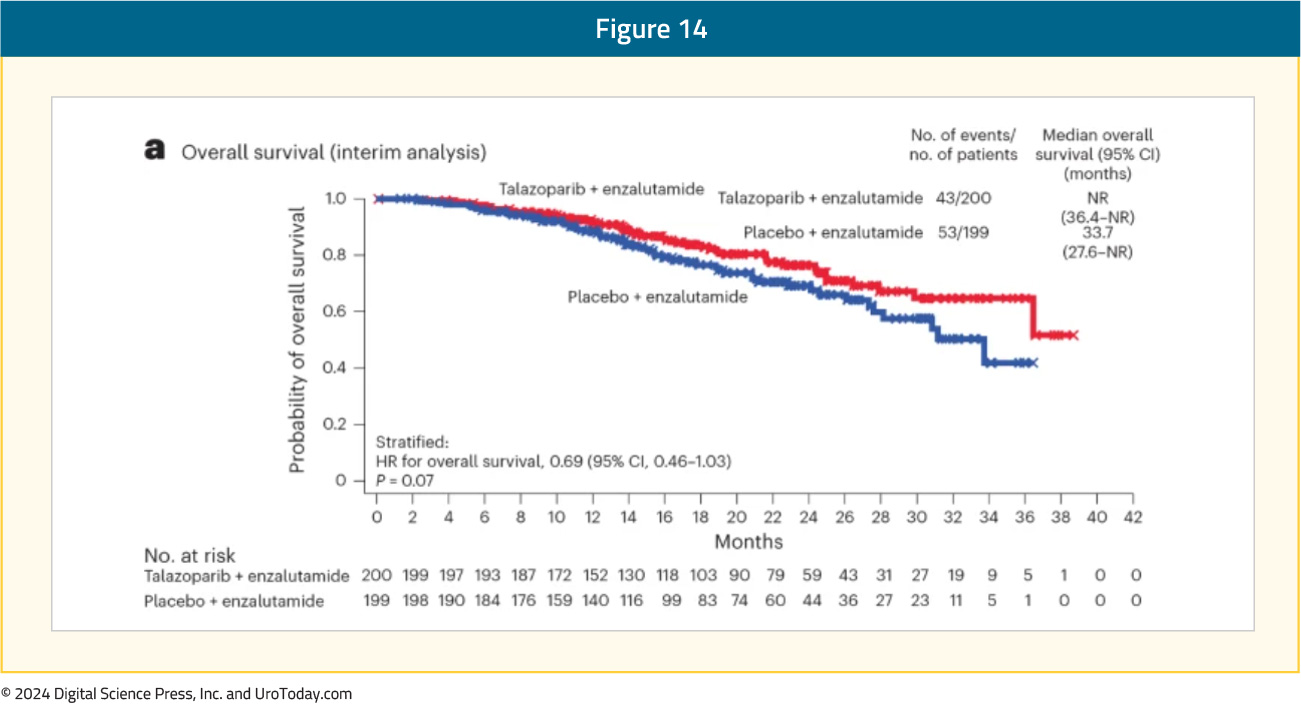
Implications for Clinical Practice
These combinations add to the growing arsenal of approved agents in the mCRPC space, which is of utmost significance given the historically poor survival outcomes of this high-risk cohort (2–3 years). There are however important considerations for the use of these combinations in contemporary clinical practice.Current guidelines strongly recommend treatment intensification in the castrate-sensitive setting with either doublet (ADT + ARPI) or triplet regimens (ADT + ARPI + docetaxel).13 While treatment intensification in the real-world setting remains subpar, future mCRPC patients are increasingly more likely to have ‘seen’ an ARPI in the castrate-sensitive setting, which contrasts with the ‘historical’ mCRPC patients enrolled in PROpel, MAGNITUDE, and TALAPRO-2.
Additionally, the utilization of such agents in clinic practice is contingent upon the identification of qualifying mutations. The majority of patients in the real world do not undergo genetic testing,14,15 and there remain practical challenges to genetic testing. The best method for detecting an HRR deficiency has yet to be determined. Although obtaining genetic testing from a fresh metastatic tumor biopsy is considered optimal, it is often unavailable and can be technically challenging to obtain in select metastatic cases (e.g., bone biopsy samples). Use of circulating tumor DNA remains an option in this setting, along with an archival sample of metastatic or primary tumor tissue, although limitations in this scenario include longitudinal accumulation of additional somatic mutations that are not present in this older sample, multifocal/multiclonal disease not representative of other tumor sites, potential degradation of DNA after years in paraffin, and potential false positive results secondary to clonal hematopoiesis of indeterminate potential (CHIP). Reliance on blood or saliva samples for germline testing does not evaluate for somatic alterations and cannot discern monoallelic from biallelic alterations, missing approximately half of actionable BRCA1/2 alterations. It thus becomes clear that clinical implementation strategies will be of critical importance to facilitate the increased utilization of these agents in practice.
Conclusions and Future Directions
The combination of PARP inhibitors and ARPIs has emerged as an FDA-approved 1st line treatment option for patients with mCRPC and BRCA1/2 mutations (abiraterone acetate/prednisone + olaparib or niraparib) or HRR mutations (talazoparib + enzalutamide). Future trials will evaluate these combinations in the metastatic hormone sensitive setting:- TALAPRO-3 (NCT04821622): Phase 3 randomized trial of talazoparib + enzalutamide versus placebo + enzalutamide in metastatic hormone sensitive prostate cancer (mHSPC) patients with DNA damage repair (DDR) mutations
- PROact (NCT05167175): Single arm phase II trial of olaparib + abiraterone acetate/prednisone in mHSPC patients
- AMPLITUDE (NCT04497844): Phase 3 randomized trial of niraparib + abiraterone acetate/prednisone versus placebo + abiraterone in HRR-mutated mHSPC patients
Published April 2024