First-line Treatment for Metastatic Castrate-resistant Prostate Cancer
Regardless of the treatment given, approximately 20%-30% of patients with localized PCa progress to metastatic disease, commonly treated with hormonal therapy.5 This can be given through surgical castration (bilateral orchiectomy) or through medical castration using androgen deprivation therapy (ADT). Both methods achieve a castrate level of serum testosterone which is regarded as the standard of care for treating metastatic hormone-sensitive PCa (mHSPC). However, mHSPC is destined to progress to metastatic castrate-resistant prostate cancer (mCRPC).6 The castrate-resistant prostate cancer (CRPC) state is defined as disease progression despite reaching castrate testosterone levels (serum testosterone < 50 ng/dL or 1.7 nmol/L), and can present as either a continuous rise in serum PSA levels, progression of pre-existing disease, and/or the appearance of new metastases.7 CRPC has a median survival of approximately three years8 and is associated with a significant deterioration of quality of life.9 The exact mechanism of transition from mHSPC to mCRPC is still unclear. However, it is known that despite castrate levels of androgens, the androgen receptor (AR) remains active and continues to drive PCa progression in CRPC.10 This has led to the development of novel agents aimed at further decreasing androgen production or blocking AR function. However, there are other biologic pathways that function independently of androgen signaling and also result in CRPC.
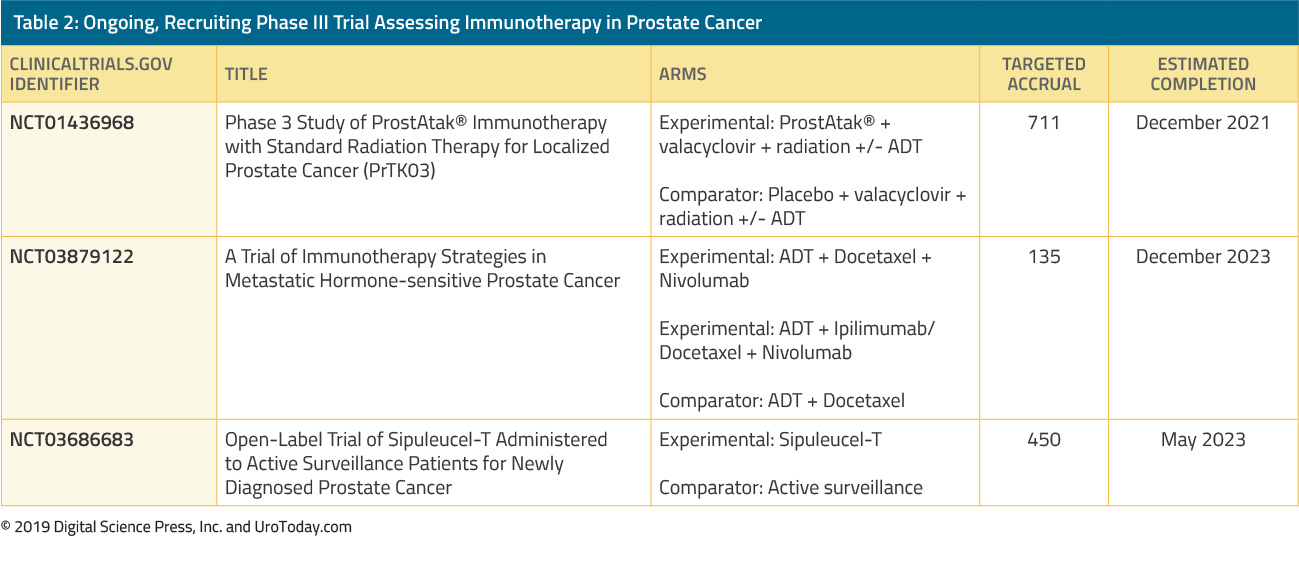
Several significant shifts have occurred in the treatment options of the mHSPC space resulting in substantial survival benefit (please see “The rapidly evolving management strategy of metastatic Hormone-Sensitive Prostate Cancer” link), including the introduction of chemotherapy in the CHAARTED study11 and STAMPEDE trial,12 the addition of abiraterone acetate and prednisone in the LATITUDE study13 and STAMPEDE trial,14 the addition of enzalutamide in the ARCHES trial15 and the ENZAMET study,16 and lastly, the addition of apalutamide, an oral nonsteroidal anti-androgen, which like enzalutamide, binds directly to the ligand-binding domain of the AR and prevents AR translocation, DNA binding, and AR-mediated transcription.17 The TITAN trial showed overall survival (OS) benefit in apalutamide-treated mHSPC patients.18 Apalutamide has also shown benefit over placebo in the non-metastatic CRPC (nmCRP) setting in the SPARTAN phase 3 placebo-controlled trial,19 similar to the benefit shown by enzalutamide-treated nonmetastatic castrate-resistant prostate cancer (nmCRPC) patients, in the PROSPER trial20 (please see “The novel treatments for the non-metastatic castrate-resistant prostate cancer” link). These treatment advances in the mHSPC and nmCRPC setting have raised the question of which treatment options should be offered to patients in the mCRPC setting.21
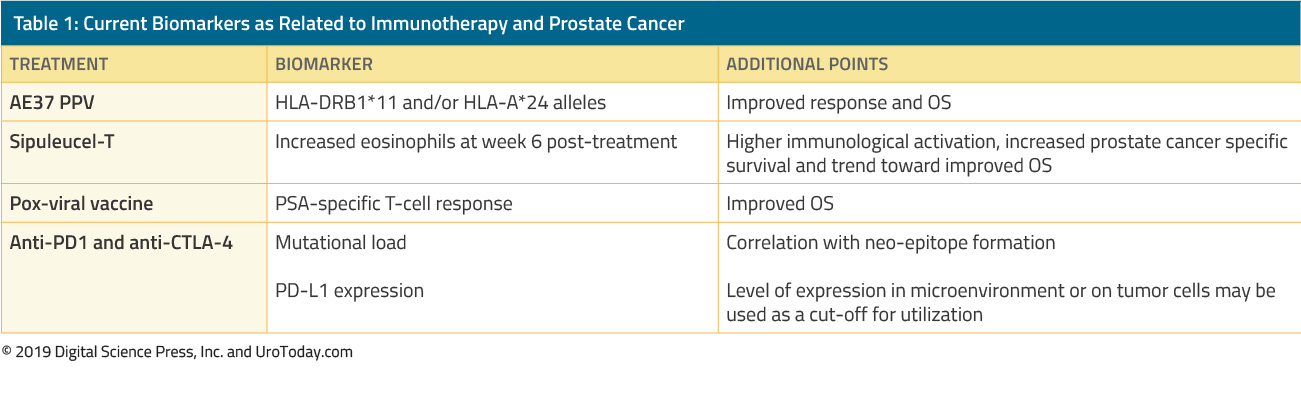
The treatment of men with CRPC has dramatically changed over the last 15 years. Prior to 2004, when patients failed primary ADT, treatments were administered solely for palliation. The landmark trials by Tannock et al.22 and Petrylak et al.23 in 2004 were the first to introduce docetaxel chemotherapy in mCRPC patients that were shown to improve their survival. However, since docetaxel was FDA approved, five additional beneficial agents showing a survival advantage have been FDA-approved based on randomized clinical trials (Table 1). These include enzalutamide, and abiraterone, which specifically affect the androgen axis, sipuleucel-T, which stimulates the immune system;24 cabazitaxel, which is another chemotherapeutic agent;25 and radium-223, a radionuclide therapy.26 Other treatments for mCRPC have shown to improve outcomes but have yet been approved by the FDA and will be discussed in another review. Due to the substantial increase in multiple FDA-approved therapeutic agents in patients with CRPC, clinicians are challenged with a plethora of treatment options and multitude potential sequences of these agents, making clinical decision-making in mCRPC significantly more complex.
Table 1. Agents that have been approved for the treatment of metastatic castrate-resistant prostate cancer in the US

mCRPC is usually a debilitating disease, and patients will most likely benefit from a management strategy formalized by a multidisciplinary team consisting of urologists, medical oncologists, radiation oncologists, nurses, psychologists, and social workers.28 It is imperative to discuss palliation treatment options when considering additional systemic treatment, including management of pain, constipation, anorexia, nausea, depression, and fatigue.
Another crucial point to consider when establishing the appropriate treatment sequence in this disease space is the associated cost. Using models that included additional lines of treatment before or after docetaxel, the mean cost of mCRPC treatment during a mean period of 28.1 months was approximately $48,000 per patient.29 This cost is quite high due to the fact that patients may receive multiple lines of therapy and incur ongoing medical services during the course of their disease.30
Only two trials have demonstrated a marginal survival benefit for patients remaining on LHRH analogs instead of adding second- and third-line therapies.31, 32 Studies have shown that CRPC is not resistant to ADT, but rather hypersensitive to it.10 Treatment-mediated selection pressure during ADT causes the AR to amplify, and ensure the situation does not escalate, ADT is continued to be administered in the mCRPC setting. Treatment-mediated selection pressure also continues throughout the entire lifespan of the tumor, intensifying the need to correctly sequence therapies. However, because prospective data are lacking, the minute potential benefit of continuing castration still outweighs the minimal risk of this treatment. In addition, all subsequently approved treatments have been studied in men with ongoing ADT, adding another reason why it should be continued.
Before delving into the actual available treatment options, it is important to recognize that it is still unclear when to begin therapy in mCRPC patients who are completely asymptomatic. It is still unknown whether earlier treatment is superior, or if we should wait until the patient becomes symptomatic and develops pain. Before starting treatment, we should consider the patient’s existing comorbidities and expected adverse effects of starting therapy. Patients with early-stage mCRPC in the COU-AA-302 trial who received abiraterone typically survived almost one year longer than those who received placebo (median OS, 53.6 months vs. 41.8 months, respectively, HR, 0.61; 95% CI, 0.43 to 0.87; P = .006).33 Thus, early-stage mCRPC patients benefited from earlier start of abiraterone. In the same trial patients with asymptomatic or mildly symptomatic mCRPC, with baseline PSA < 15.6 ng/mL abiraterone also led to a faster rate and a greater degree of PSA decline than placebo.34 Although the currently available data is limited, it most likely suggests that starting treatment earlier rather than later is more advantageous.33, 34
Approved first-line treatment options for metastatic castrate-resistant prostate cancer
Abiraterone
Abiraterone is an antiandrogen which is an inhibitor of 17α-hydroxylase/C17,20-lyase (CYP17) enzyme. The COU-AA-302 phase III study evaluated abiraterone in 1,088 chemo-naïve, asymptomatic or mildly symptomatic mCRPC patients without visceral metastases. In this trial patients were randomized to abiraterone acetate or placebo, both combined with prednisone35 (Figure 1). Patients were stratified by either the Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 and by asymptomatic or mildly symptomatic disease.35 OS and radiographic progression-free survival (rPFS) was the co-primary end-points. The trial demonstrated that after a median follow-up of 22.2 months, there was a significant improvement of rPFS in the abiraterone arm (median 16.5 vs. 8.2 months, HR 0.52, p < 0.001). At the final analysis after a median follow-up of 49.2 months, the OS end-point was significantly positive (34.7 vs. 30.3 months, HR: 0.81, 95% CI: 0.70-0.93, p = 0.0033).36 It is important to remember that mCRPC spans a broad prognostic spectrum even when it is chemotherapy-naïve.37 In an analysis of the abiraterone arm of the COU-AA-302 study, patients who had no pain at baseline, normal alkaline phosphatase and LDH levels, and less than 10 bone metastases had a median OS of 42.6 months.37 However, patients with more risk factors for progression had significantly shorter median OS.37 When assessing the toxicity profile of abiraterone, it seemed to confer more adverse events related to mineralocorticoid excess and liver function abnormalities, but these were mostly graded 1-2 adverse effects. Lastly, abiraterone was also shown to be equally effective in the elderly population (> 75 years).38
Figure 1. COU-AA-302 study design
Enzalutamide
Enzalutamide is a nonsteroidal antiandrogen. The PREVAIL study which is a randomized phase III trial included 1,717 chemo-naïve mCRPC patients and patients with visceral metastases were eligible as well.39 This trial compared enzalutamide to placebo (Figure 2). The PREVAIL trial showed a significant improvement in enzalutamide-treated patients in both co-primary endpoints, which included rPFS (HR: 0.186; CI: 0.15-0.23, p < 0.0001), and OS (HR: 0.706; CI: 0.6-0.84, p < 0.001). Extended follow-up and final analysis confirmed a benefit in OS and rPFS for enzalutamide.40 In 78% of patients treated with enzalutamide a PSA decrease of more than 50% was reported. The most common clinically relevant adverse events were fatigue and hypertension. Enzalutamide was also equally effective and well-tolerated in older men (> 75 years)41 and in those with or without visceral metastases.42 However, for men with liver metastases, there seemed to be no discernible benefit.43 The TERRAIN trial compared enzalutamide with bicalutamide, an older antiandrogen, in a randomized double-blind phase II study, showing a significant improvement in PFS (15.7 months vs. 5.8 months, HR: 0.44, p < 0.0001) in favor of enzalutamide.44
Figure 2. PREVAIL study design
Docetaxel
The landmark trial TAX 327 showed a significant improvement in median OS of 2-2.9 months in mCRPC patients treated with docetaxel-based chemotherapy when compared to patients who were treated with mitoxantrone plus prednisone therapy.22 The SWOG 9916 trial compared mitoxantrone to docetaxel and showed similar results23 (Figure 3). The standard first-line chemotherapy is docetaxel 75 mg/m2 in three-weekly doses combined with prednisone 5 mg twice a day, up to ten cycles. There are several important prognostic factors to consider when administering docetaxel: visceral metastases, pain, anemia (Hb < 13 g/dL), bone scan progression, and prior estramustine therapy. These prognostic factors may help to stratify response to docetaxel. Using these prognostic factors the disease can be categorized into low, intermediate and high risk, with significantly different corresponding median OS estimates of 25.7, 18.7 and 12.8 months, respectively.45 Although age by itself is not a contraindication to docetaxel therapy, patients must be fit enough to endure this type of treatment and comorbidities should be assessed prior to treatment initiation. In men who are thought to be unable to tolerate the standard dose and schedule of docetaxel, this can be decreased from 75 to 50 mg/m2 every two weeks, showing less grade 3-4 adverse events and a longer time to treatment failure.46
Figure 3. SWOG 9916 and TAX 327 trial designs
Sipuleucel-T
Sipuleucel-T, an autologous active cellular immunotherapy, was shown in a phase III trial (IMPACT trial) to confer a survival benefit in 512 asymptomatic or minimally symptomatic mCRPC patients when compared to placebo24 (Figure 4). After a median follow-up of 34 months, the median survival was significantly higher in the sipuleucel-T group (25.8 vs. 21.7 months, with an HR of 0.78,p = 0.03).24 Importantly, no PSA decline was observed during or after treatment and PFS was similar in both arms. The overall tolerance to sipuleucel-T was very good, with mostly grade 1-2 adverse events occurring. Currently, this treatment is only available in the US and is no longer available in Europe.
Figure 4. IMPACT trial design
Conclusions
In the last 15 years, there has been considerable scientific progress and investment in drug development for patients with mCRPC. This has resulted in the FDA approval of several lines of systemic therapies on grounds of pain palliation, minimizing disease adverse effects, and OS prolongation. To date, the reported impact on OS in mCRPC patients from each of these individual agents is still modest, resulting in an addition of only a few months. It is necessary to enhance our understanding of the disease biology of mCRPC, integrate a comprehensive molecular understanding of castration resistance, and analyze mechanisms of resistance to current therapies to improve future treatment development. It is also crucial to invest and develop predictive biomarkers to assist in the personalization of therapy. Lastly, on a more practical note, more data is needed on the appropriate second and third-line therapies, and sequencing and combination of available medications, discussed in more detail in the next review article (“Beyond first line treatment of metastatic castrate-resistant prostate cancer”).Published Date: November 19th, 2019